---
description: "Matrices de proyección poblacional bayesianas (MPP bayesianas) que incorporan incertidumbre en transiciones y fecundidad. Implementación en R con priors informativos."
image: "images/Lepanthes_eltoroensis_Edwin_Guevara.jpg"
open-graph:
description: "Matrices de proyección poblacional bayesianas (MPP bayesianas) que incorporan incertidumbre en transiciones y fecundidad. Implementación en R con priors informativos."
twitter-card:
description: "Matrices de proyección poblacional bayesianas (MPP bayesianas) que incorporan incertidumbre en transiciones y fecundidad. Implementación en R con priors informativos."
---
# Acercamiento bayesiano para calcular transiciones y fecundidades
Por: Raymond L. Tremblay
Los modelos poblacionales bayesianos permiten integrar explícitamente la incertidumbre demográfica, de muestreo y de proceso en el análisis de la dinámica poblacional. Este capítulo introduce las matrices de proyección poblacional bayesianas (MPP bayesianas) como una extensión coherente de los modelos matriciales clásicos.
## Matrices de proyección poblacional bayesianas: integrar incertidumbre y proceso
Las matrices de proyección poblacional bayesianas (MPP bayesianas) extienden los modelos matriciales tradicionales al tratar los parámetros vitales (supervivencia, crecimiento y fecundidad) como variables aleatorias en lugar de valores puntuales fijos. Esta formulación permite propagar explícitamente la incertidumbre desde los datos primarios hasta las proyecciones de abundancia y crecimiento poblacional ($\lambda$), algo fundamental en poblaciones con tamaños de muestra reducidos, series temporales cortas o alta variabilidad ambiental.
En este capítulo se presenta el marco conceptual de las MPP bayesianas, mostrando cómo se combinan modelos de observación y proceso dentro de una jerarquía bayesiana para estimar matrices poblacionales con distribuciones posteriores completas. Se discuten las ventajas clave de este enfoque frente a métodos frecuentistas, incluyendo la incorporación de información previa, el manejo coherente de datos faltantes y la cuantificación directa de la incertidumbre en predicciones a corto y largo plazo.
A través de ejemplos aplicados, se ilustra cómo las MPP bayesianas facilitan la evaluación del riesgo poblacional, la comparación entre escenarios de manejo y la integración posterior con análisis de elasticidad, sensibilidad y LTRE. Este capítulo se diferencia de los anteriores porque desplaza el énfasis desde la estimación puntual de parámetros hacia la inferencia probabilística completa de la dinámica poblacional, un enfoque especialmente relevante en biología de la conservación y ecología aplicada.
```{r, message=FALSE, warning=FALSE}
#| docx_fit: true
#| label: fig-bayes-setup
#| fig-cap: "Carga de los paquetes necesarios (tidyverse, popbio, raretrans, DiagrammeR, Rage, flextable) y de las funciones auxiliares para generar las figuras."
if (!require("pacman")) install.packages("pacman")
pacman::p_load(janitor, tidyverse)
library(tidyverse)
library(janitor)
library(popbio) # para la función projection.matrix()
library(raretrans) # disponible en CRAN: install.packages("raretrans")
library(DiagrammeR)
library(Rage)
library(flextable)
source("R/figuras_helpers.R") # plc_save(), grviz_save(), save_plot_png()
```
```{r bayes-theme}
source("R/rlt_theme.R") # tema + paleta canónicos (R/rlt_theme.R)
```
## ¿Qué es un análisis bayesiano? Una introducción rápida
En estadística clásica (frecuentista), tratamos los parámetros (e.g., la probabilidad de transición de plántula a juvenil) como **valores fijos pero desconocidos**, y los datos como variables aleatorias. La inferencia consiste en estimar el parámetro a partir de los datos: si observamos 2 transiciones de 7 plántulas, el estimador puntual es 2/7 = 0.286, sin más información.
En el marco bayesiano, invertimos la perspectiva: los **datos son lo que observamos**, y los **parámetros se tratan como variables aleatorias** sobre las que tenemos creencias. Esas creencias se expresan como una **distribución previa** (lo que sabíamos antes de mirar los datos) y se actualizan a una **distribución posterior** (lo que sabemos después). La regla de actualización es la fórmula de Bayes:
$$\underbrace{P(\theta \mid \text{datos})}_{\text{posterior}} \propto \underbrace{P(\text{datos} \mid \theta)}_{\text{verosimilitud}} \times \underbrace{P(\theta)}_{\text{previa}}$$
En español plano: la posterior es proporcional a la previa multiplicada por la verosimilitud. La previa aporta información cuando los datos son escasos; la verosimilitud (los datos) domina cuando hay muchas observaciones. **Esta propiedad es exactamente lo que necesitamos para especies raras**: si observamos 2 plántulas y 0 transiciones a juvenil, el estimador frecuentista da $\hat{p} = 0$ (lo cual implica imposibilidad biológica), mientras que el estimador bayesiano combina las 2 observaciones con la previa y da una probabilidad pequeña pero positiva, con incertidumbre cuantificada.
::: callout-note
**La regla de Bayes en lenguaje cotidiano.**
$$\text{Lo que crees ahora} \;\propto\; \text{Lo que creías antes} \;\times\; \text{Lo que dicen los datos}$$
Cuando los datos son muchos y consistentes, dominan: la posterior se parece a la verosimilitud. Cuando los datos son escasos o ambiguos, la previa importa: la posterior se parece a la previa. El método bayesiano hace este equilibrio explícito y reproducible.
:::
::: callout-tip
**Por qué leer este capítulo (incluso si nunca has hecho un análisis bayesiano).** La estimación clásica de matrices de transición —`projection.matrix()`— es perfectamente adecuada cuando el tamaño de muestra es grande y cada transición se observó muchas veces. Pero en estudios reales de orquídeas, casi nunca es así: las muestras son pequeñas, varias transiciones tienen 0 observaciones, y el resultado es una matriz con ceros que la biología sabe que no son cero. El enfoque bayesiano resuelve esto sin "inventar" datos: usa explícitamente lo que ya sabes sobre el ciclo de vida (la previa) y produce estimaciones realistas con intervalos de credibilidad. Aprender a usarlo es una de las inversiones de mayor retorno en la dinámica poblacional aplicada.
:::
## Introducción a las MPP bayesianas
Este capítulo presenta una alternativa bayesiana para estimar los parámetros de las matrices de transición y fecundidad en modelos poblacionales, especialmente útil en el estudio de especies raras. Estas especies suelen tener tamaños de muestra reducidos o transiciones vitales poco frecuentes, lo que dificulta obtener estimaciones confiables. El enfoque bayesiano propuesto permite incorporar conocimiento previo sobre el ciclo de vida de la especie para generar estimaciones más realistas.
### Ejemplo de especie con tres etapas
Consideremos una especie hipotética con tres etapas en su ciclo de vida: semillas, plántulas y adultos. En este caso, sólo los adultos producen semillas. El primer paso del análisis consiste en identificar qué etapas tienen mayor influencia en la dinámica poblacional y cómo optimizar la recolección de datos en campo.
La primera figura muestra el ciclo de vida considerado. Tras definirlo, se recolectaron datos de campo y se estimaron las tasas de transición y fecundidad. Los resultados revelaron lo siguiente:
1. **No se observaron semillas**. Esto podría indicar que todas germinaron y pasaron a la etapa de plántula.
- ¿Es realista asumir que el 100% de las semillas germinaron?
- ¿Por qué no se encontraron semillas en el muestreo?
2. **No se observaron plántulas**. Esto podría interpretarse como que todas murieron o que ninguna semilla germinó.
- ¿Es realista asumir que todas las plántulas mueren durante el ciclo de vida de una especie?
Estas interpretaciones extremas —que ningún individuo muere o que todos mueren— no son biológicamente realistas. Sin embargo, pueden surgir como resultado de un tamaño de muestra reducido. Por ejemplo, si sólo se observaron 4 plantas adultas (debido a que la especie es rara) y todas sobrevivieron, se estimaría una tasa de supervivencia del 100%, lo que implicaría que los individuos son "inmortales". Este resultado no refleja la realidad biológica, sino una limitación del muestreo: por simple azar, no se observó ninguna muerte. Por lo tanto, es fundamental reconocer cuándo los parámetros estimados no representan adecuadamente la historia de vida típica de la especie.
### El papel del paquete `raretrans`
El paquete `raretrans` fue desarrollado para abordar este tipo de situaciones. Utiliza un enfoque bayesiano que permite incorporar conocimiento previo sobre el ciclo de vida de la especie, ayudando a estimar parámetros más realistas incluso cuando los datos son escasos o incompletos.
### Ejemplos de interpretaciones no realistas
Algunas interpretaciones biológicamente poco plausibles que pueden surgir del análisis de datos limitados incluyen:
- Todas las semillas germinan y se convierten en plántulas.
- Ninguna plántula sobrevive ni alcanza la etapa adulta.
- Todos los adultos sobreviven sin mortalidad.
- Ningún adulto produce semillas.
**Nota**: Aunque estos resultados pueden surgir directamente de los datos recolectados, muchas veces son consecuencia de tamaños de muestra pequeños o de eventos raros que no se observaron durante el periodo de muestreo. Por ello, es esencial considerar tanto la biología de la especie como las limitaciones del muestreo al interpretar los datos.
```{r bayes1, message=FALSE}
matA <- rbind(
c(0.0, 0.0, 0.0),
c(1.0, 0.0, 0.0),
c(0.0, 0.0, 0.9)
)
stages <- c("semillas", "plantulas", "adultos")
title <- NULL
```
## Evaluación crítica del ciclo de vida y la matriz poblacional
El ciclo de vida propuesto para esta especie hipotética presenta limitaciones importantes que lo hacen poco realista. En particular:
- No se observaron semillas en el muestreo, lo que sugiere que todas germinaron y pasaron a la etapa de plántula. Sin embargo, asumir una germinación del 100% es altamente improbable en condiciones naturales.
- Ninguna plántula sobrevivió, lo que indica una falla completa en la transición hacia la etapa adulta.
Como resultado, la tasa de crecimiento poblacional asintótica observada es $\lambda = 0.9$ lo que implica una población en declive.
Además, la matriz poblacional estimada presenta dos propiedades estructurales problemáticas:
1. **No es ergódica**: No es posible transitar desde cualquier estado a cualquier otro estado dentro del ciclo de vida. Esto limita la conectividad entre etapas y reduce la capacidad de la matriz para representar adecuadamente la dinámica poblacional.
2. **Es reducible**: Una o más filas y columnas pueden eliminarse sin alterar las propiedades fundamentales de la matriz (como sus eigenvalores y eigenvectores). Esto indica que ciertas etapas del ciclo de vida no contribuyen significativamente a la dinámica poblacional, lo cual puede ser un artefacto del muestreo o de una estructura biológica mal representada.
```{r}
#| docx_fit: true
#| label: fig-bayes2
#| fig-cap: "Diagrama del ciclo de vida correspondiente a la matriz de proyección matA con sus estadios y transiciones."
plc_save(matA, stages = stages, png = "images/bayes2.png")
```
### Interpretaciones biológicamente irreales de los datos existentes
Al estudiar especies longevas como *Sequoia sempervirens*, es común enfrentar interpretaciones erróneas derivadas de datos limitados o mal contextualizados. Esta especie puede alcanzar tamaños extraordinarios (diámetros a la altura del pecho, *dbh*, mayores a 3 metros) y tener una longevidad estimada entre 1,200 y 2,200 años.
Supongamos que se realiza un muestreo de más de 1,000 individuos grandes. Es posible que, en un intervalo de uno o incluso diez años, no se observe la muerte de ningún individuo. A partir de estos datos, se podría concluir erróneamente que la supervivencia anual es del 100%. Sin embargo, esta interpretación es biológicamente poco realista por varias razones:
1. **Tamaño de muestra insuficiente**: Si el muestreo se ampliara a 10,000 o 100,000 individuos, probablemente se detectarían muertes, aunque fueran pocas. Esto sugiere que la mortalidad es baja, pero no nula.
2. **Relación entre tamaño y mortalidad**: En plantas, la probabilidad de morir suele disminuir con el tamaño del individuo. Los árboles más grandes tienen mayor resistencia a factores ambientales y biológicos, lo que reduce su tasa de mortalidad.
3. **Dependencia temporal de la mortalidad**: La muerte de individuos puede estar influenciada por eventos bióticos (plagas, enfermedades) o abióticos (sequías, incendios, tormentas) que varían entre años. En años extremos, la mortalidad puede aumentar significativamente.
Por lo tanto, aunque los datos observados sugieren una supervivencia perfecta, la probabilidad real de mortalidad es baja pero no cero. Este tipo de sesgo en la interpretación puede ser corregido mediante enfoques estadísticos más robustos, como el uso de modelos bayesianos que incorporan incertidumbre y conocimiento previo sobre la biología de la especie.
------------------------------------------------------------------------
## El paquete `raretrans`
En la siguiente sección usaremos el paquete `raretrans`, que facilita los estimados de las entradas en las matrices de transición y fecundidades. El manuscrito [@tremblay2021population; @raretrans2024] contiene los datos originales y el uso del paquete. Para tener acceso a los documentos sigan el siguiente enlace: <https://doi.org/10.1016/j.ecolmodel.2021.109526>.
**Population projections from holey matrices: Using prior information to estimate rare transition events**
Resumen
Las matrices de proyección poblacional (MPP) son una herramienta común para predecir el comportamiento de la población a corto y largo plazo. Sin embargo, cuando se utilizan para especies raras, amenazadas y en peligro de extinción los datos disponibles suelen tener tamaños de muestra reducido. En consecuencia, algunos eventos demográficos podrían perderse en la modelización dando como resultado matrices con eventos faltantes que reflejen trayectorias de ciclo de vida incompletas o biológicamente poco realistas. Estas matrices con eventos faltantes (ceros; e.g., sin observación de semillas que se transforman en plántulas) a menudo se sustituyen utilizando información de la literatura aunque información referente a otras poblaciones, otros períodos de tiempo, otras especies usualmente se omite.
Una forma de abordar este problema es a través de métodos bayesianos para el ajuste de los parámetros matriciales. Este marco estadístico nos permite usar información previa para completar los valores faltantes usando, por ejemplo, una distribución de Dirichlet para incluir información sobre las transiciones de estado y una distribución Gamma como previa para la fecundidad. Este método permite integrar información conocida (*a priori*) e incluir explícitamente el peso de la información previa en las distribuciones posteriores. Además, al establecer explícitamente las distribuciones previas, los resultados son reproducibles y se pueden volver a evaluar con diferentes distribuciones *a priori* en el futuro. A continuación mostramos cómo el uso de estos métodos en datos reales influye en la dispersión de los resultados posteriores y origina matrices irreducibles y ergódicas, permitiendo hacer inferencias biológicamente más realistas sobre las probabilidades de transición y fecundidad.
## Instalación del paquete raretrans
El paquete `raretrans` ya está disponible en CRAN, por lo que se puede instalar de la manera habitual. Solo es necesario ejecutar la instrucción una vez; en ejecuciones posteriores basta con cargar la biblioteca con `library(raretrans)`.
```{r bayes4, message=FALSE}
#| eval: false
# Instalar desde CRAN (ejecutar solo una vez)
install.packages("raretrans")
```
```{r bayes4b, message=FALSE}
library(raretrans)
```
El contenido que se presenta a continuación es una traducción y ampliación de la documentación del siguiente enlace:
<https://atyre2.github.io/raretrans/articles/onepopperiod.html>
A continuación se ejemplifica el uso del paquete `raretrans` para estimar los parámetros en una población y en un periodo de transición determinados.
```{r bayes5, message = FALSE}
# Mi tema de formato de las gráficas de ggplot2 personal
rlt_theme <- theme(
text = element_text(family = "Libertinus Serif"),
axis.title.y = element_text(colour = "grey20", size = 15, face = "bold"),
axis.text.x = element_text(colour = "grey20", size = 15, face = "bold"),
axis.text.y = element_text(colour = "grey20", size = 15, face = "bold"),
axis.title.x = element_text(colour = "grey20", size = 15, face = "bold")
)
```
## Obtención de la matriz de proyección
`raretrans` supone que la matriz de proyección es una lista con dos componentes:
- **Matriz de transición**: Probabilidades de pasar de una etapa a otra (o permanecer en la misma).
- **Matriz de fecundidad**: Número de nuevos individuos producidos por cada etapa reproductiva.
Este es el formato de salida de `popbio::projection.matrix()`. Si tenemos transiciones individuales en nuestra base de datos (con clase `data.frame()`), podemos usar `raretrans::get_state_vector()` para obtener el número inicial de individuos por etapa.
Podemos utilizar la función `popbio::projection.matrix()` para obtener los datos necesarios. Hacemos una demostración con los datos de transición y fecundidad de *Lepanthes eltoroensis* **POPNUM 250** en el **periodo 5**. *Lepanthes eltoroensis* es una orquídea epífita, endémica de Puerto Rico y con una distribución limitada a una pequeña región de la isla.
::: {style="display: flex; justify-content: center; gap: 10px;"}
{#fig-lepanthes-eltoroensis-edwin-guevara}
{#fig-lepanthes-eltoroensis-phorophyte-edwin-guevara}
:::
------------------------------------------------------------------------
<br>
## Descargar los datos de una población de *L. eltoroensis* del paquete `raretrans`
```{r bayes6}
data("L_elto") # carga el conjunto de datos `L_elto` en la memoria de la computadora (los datos están incluido en el paquete `raretrans`)
head(L_elto) |> flextable()
```
## Organización de los datos en el "data.frame"
Supongamos que tienes una base de datos llamada `L_elto` con las siguientes columnas:
- `pop`: identificador de la población
- `period`: periodo de tiempo
- `stage`: etapa actual del individuo
- `next_stage`: etapa siguiente
- `fertility`: indicador de fecundidad
Seleccionamos la población POPNUM 250 y el periodo 5:
El primer paso es seleccionar los datos de la población y el periodo de tiempo que queremos analizar. Posteriormente, cambiamos el nombre del estado más pequeño a "seedling". Este paso no es estrictamente necesario pero nos ayuda a entender más fácilmente la nomenclatura.
La base de datos `L_elto` está compuesta por datos individuales del estado actual (`stage`, periodo **t**), del estado siguiente (`next_stage`, periodo **t+1**) y de la fertilidad. Tenga en cuenta que aquí "p" significa "plántula" por la traducción al español. En la siguiente caja de código las primeras líneas seleccionan la población de interés y el periodo de tiempo sobre el que queremos trabajar y cambian el nombre de la etapa del ciclo vital de "p" a "s".
```{r bayes7_mungeData}
onepop <- L_elto %>%
filter(POPNUM == 250, year == 5) %>% # Filtrar la población # 250, el periodo (año=year) 5
mutate(
stage = case_when(stage == "p" ~ "s", TRUE ~ stage),
next_stage = case_when(next_stage == "p" ~ "s", TRUE ~ next_stage)
) # redefine "p" por plantula a "s" para seedling
```
En el siguiente código usamos la función `popbio::projection.matrix()` para crear la matriz de proyección poblacional. Tenemos ahora nuestra matriz de transición y fecundidad basadas en los datos de la población #250 del periodo 5. La función `projection.matrix()` requiere que se especifiquen los nombres de las etapas y los estados siguientes, así como el nombre de la columna de fecundidad. En este caso, las etapas son "s" (plántula), "j" (juvenil) y "a" (adulto), y el estado siguiente es `next_stage`. La columna de fecundidad se llama `fertility`. El argumento `sort` especifica el orden en que queremos que aparezcan las etapas en la matriz.
Solamente los adultos pueden producir plántulas, por lo que la matriz de fecundidad tiene un valor solamente en la posición (1,3), (plántula, adulto). La función `projection.matrix()` devuelve una lista con dos matrices: una de transición y otra de fecundidad. La matriz de transición es una matriz cuadrada que muestra las probabilidades de transición entre las etapas, mientras que la matriz de fecundidad es una matriz que muestra la fecundidad por etapa.
A partir de este punto los análisis se llevan a cabo considerando **s** (plántula), **j** (juvenil) y **a** (adulto), y las dos matrices: **T** (transición de estadios) y **F** (fecundidad). La tasa de crecimiento asintótica de la población observada es $\lambda$. Las transiciones raras que faltan en nuestra primera matriz de transición `TF$T`, pero que sabemos que ocurren, son la transición de plántula (*s*) a adulto (*a*) y la transición de *j* a *s*.
```{r bayes8_projectionMatrix, message=FALSE}
# popbio::projection.matrix no funciona con el formato *tibbles*, por consecuencia se convierte en data.frame
head(onepop) |> flextable() # Ahora tenemos solamente datos de la población #250 del periodo 5
# Crear TF = TRUE, añadir para formatear correctamente.
TF <- popbio::projection.matrix(
as.data.frame(onepop),
stage = stage,
fate = next_stage,
fertility = "fertility",
sort = c("s", "j", "a"),
TF = TRUE
)
TF # Esta es la estructura de etapas de vida para esa población. Nota que tenemos dos matrices, una de transiciones **T** y otra de fecundidad **F**.
```
------------------------------------------------------------------------
## Obtener el número inicial de individuos por etapa
Dado que nuestros conteos (número de individuos), $N$, y el tamaño de *muestra equivalente* *a priori* se expresan como múltiplos del número de individuos observados, necesitamos obtener el número de individuos en cada etapa ($N$) en el primer periodo de tiempo. Para esto utilizamos la función `raretrans::get_state_vector()`. Se observa que la cantidad de individuos iniciales por etapa —es decir, el número de individuos en el primer muestreo— es muy limitada: 11 plántulas, 47 juveniles y 34 adultos para esta población en este periodo de muestreo.
```{r bayes8}
N <- get_state_vector(onepop, stage = stage, sort = c("s", "j", "a"))
N # Un vector # de individuos iniciales para cada etapa, nota que la cantidad por etapa, "stage", son los individuos en el primer muestreo
```
## Método alterno para calcular transiciones, fecundidades y el número de individuos por etapas
La lista de matrices y el vector de conteos de individuos no tienen por qué proceder de un *data frame* como hemos hecho anteriormente. Mientras tengan el formato esperado, pueden crearse a mano. Usemos la población 231 en el periodo 2 como ejemplo para dividir la matriz poblacional en las submatrices de transición **T** y de fecundidad **F**. En el siguiente código, *m* denota mortalidad, es decir, plantas que están muertas. Note que aquí las transiciones están estimadas con un tamaño de muestra todavía más pequeño: solamente 2 plántulas, 6 juveniles y 16 adultos. Ninguna de las 2 plántulas pasó a la etapa de juveniles; en consecuencia, esta transición es cero. Además, una de estas plántulas murió, por lo que la supervivencia es del 50%. Sin embargo, si hubieran muerto las dos plántulas la mortalidad sería del 100%, y si hubieran sobrevivido las dos plántulas la supervivencia sería del 100%: ¡no hay valores intermedios!
```{r bayes9}
Tmat <- L_elto %>%
mutate(
stage = factor(stage, levels = c("p", "j", "a", "m")),
fate = factor(next_stage, levels = c("p", "j", "a", "m"))
) %>%
filter(POPNUM == 231, year == 2) %>%
as.data.frame() %>%
xtabs(~ fate + stage, data = .) %>%
as.matrix()
Tmat <- Tmat[, -4] # remover la columna para transiciones de la muerte
N2 <- colSums(Tmat) # obtener el número total. Averiguar: debe ser antes de 86 en este caso.
Tmat <- sweep(Tmat[-4, ], 2, N2, "/") # normalizar a 1, descartando las transiciones *hacia* la muerte, nota que el "2" es para decir que haga los calculos por columna
# averiguar cuánta reproducción ocurrió en el año 2 mirando el año 3
# L_elto %>%
# mutate(stage = factor(stage, levels=c("p","j","a","m")),
# fate = factor(next_stage, levels=c("p","j","a","m"))) %>%
# filter(POPNUM == 231, year == 3, stage == "p") %>%
# count(stage)
# 2 descendientes desde N[3] == 16 adultos
Fmat <- matrix(0, nrow = 3, ncol = 3) # crear una matriz de ceros.
Fmat[1, 3] <- 2 / 16 # contar 16 adultos reproductivos en el tiempo t y dos plántulas en el próximo tiempo t+1, 2/16.
TF2 <- list(Tmat = Tmat, Fmat = Fmat)
```
La matriz de transición, la matriz de fecundidad y el vector de tamaño poblacional para la población 231, periodo 2, son:
```{r bayes10}
# TF2 es una lista con $Tmat (transición) y $Fmat (fecundidad).
mat_to_ft(TF2$Tmat)
mat_to_ft(TF2$Fmat)
pretty(N2) # N2 es un vector con nombres → flextable de 2 columnas
```
------------------------------------------------------------------------
### ¿Cómo se ve el ciclo de vida de la población en ese periodo?
A la matriz mostrada le falta la transición de plántula a juvenil. Además, dado que ninguno de los 6 juveniles murió, hay una sobreestimación de su supervivencia. La tasa de crecimiento asintótica de la población observada es $\lambda$. La matriz no es ergódica (no se puede llegar a cualquier otro estado desde uno o más estados), y es reducible, lo que significa que una o más columnas y filas se pueden descartar y tienen las mismas propiedades en términos de sus eigenvalores y eigenvectores.
Lo que vimos anteriormente fueron algunos de los problemas reales que encontramos cuando usamos datos de campo y hay tamaños de muestra pequeños o transiciones raras. A continuación veremos cómo resolverlos.
```{r}
#| docx_fit: true
#| out.width: "100%"
#| label: fig-bayes11
#| fig-cap: "Diagrama del ciclo de vida de la matriz de transiciones Tmat con los estadios plántulas, juveniles y adultos."
stages2 <- c("plantulas", "juveniles", "adultos")
title <- NULL
plc_save(Tmat, stages = stages2, png = "images/bayes11.png")
```
------------------------------------------------------------------------
### Incorporar previas informativas
Para evitar que el modelo genere transiciones imposibles entre etapas del ciclo de vida, se pueden incorporar previas informativas. Estas previas se basan en el conocimiento experto sobre la especie, en este caso proporcionado por un especialista en orquídeas epífitas del género *Lepanthes* (RLT). La información debe tener el mismo formato que la matriz de transición, es decir:
- Una matriz con una fila más que columnas.
- La última fila representa los individuos que mueren en cada etapa.
Es importante que estos valores se definan antes de recolectar los datos de campo, para evitar que el análisis se vea influenciado o sesgado por los datos observados.
```{r bayes12}
RLT_Tprior <- matrix(
c(
0.25, 0.025, 0.0,
0.05, 0.9, 0.025,
0.01, 0.025, 0.95,
0.69, 0.05, 0.025
),
byrow = TRUE, nrow = 4, ncol = 3
)
```
Note que en la matriz, la 1ª fila, 3ª columna es 0.0, porque esta transición es imposible. Las previas se construyen de manera que las columnas sumen 1, lo que crea mayor flexibilidad para la ponderación de los valores *a priori*. Por defecto, la suma es 1, interpretado como un *tamaño de muestra a priori* de 1.
Ahora, usando la función `fill_transitions()` se agregan los valores del campo **TF**, el tamaño de muestra en el tiempo inicial **N**, la matriz de valores con *priors* informativos, y se calcula la matriz de transición *a posteriori*. Compare **TF** con esta matriz para ver los cambios.
```{r bayes13}
fill_transitions(TF, N, P = RLT_Tprior)
```
------------------------------------------------------------------------
### Modificar el peso de los valores *a priori*
Cuando usamos previas informativas en el modelo, podemos ajustar su peso para reflejar cuánto creemos en la previa frente a los datos. Esto se hace con el argumento `priorweight` de `fill_transitions()`.
**¿Cómo funciona `priorweight` exactamente?** El argumento es un **multiplicador sobre el tamaño de muestra observado** $N$:
- La contribución efectiva de la previa = `priorweight` × $N$ (en unidades de "observaciones equivalentes").
- El tamaño de muestra efectivo total = $N$ + `priorweight` × $N$ = $N \times (1 + \text{priorweight})$.
- Por defecto `priorweight = -1`, lo que hace que la previa contribuya solamente su suma original (típicamente 1, ya que cada columna de la matriz de previas se construye para sumar 1).
```{r bayes14}
fill_transitions(TF, N, P = RLT_Tprior, priorweight = 0.5)
```
::: callout-important
**El "peso de la previa" es un tamaño de muestra equivalente.** Asignar `priorweight = 0.5` con $N = 2$ observaciones es como decir: "antes de salir al campo, mi creencia tiene la misma fuerza que $0.5 \times 2 = 1$ observación adicional". Si tu previa tiene peso efectivo 5 y observas 50 transiciones, los datos pesarán 10 veces más que la previa. La regla práctica es dejar que los datos dominen: el peso de la previa debería ser **mucho menor** que el tamaño de muestra observado, salvo que tengas razones biológicas muy fuertes para confiar en la previa.
:::
Algunos valores típicos de `priorweight`:
- `priorweight = -1` (defecto): la previa contribuye su suma original, típicamente 1 observación equivalente. Útil como previa débil.
- `priorweight = 0.1` a `0.5`: la previa pesa entre 10% y 50% del tamaño de muestra observado. Apropiado cuando hay conocimiento experto pero los datos deben dominar.
- `priorweight = 1`: la previa pesa lo mismo que los datos. Sólo justificado si los datos son muy escasos y la previa es muy confiable.
- `priorweight > 1`: la previa pesa más que los datos. Rara vez justificado en estudios poblacionales.
::: callout-warning
**Cuándo no usar una previa informativa fuerte.** Las previas informativas son apropiadas cuando hay conocimiento biológico sólido que no aparece en los datos (e.g., experiencia directa con la especie, estudios en poblaciones cercanas). **No** son apropiadas cuando:
- El tamaño de muestra ya es grande y todas las transiciones se observaron varias veces — la previa solo introduce sesgo.
- No hay base experta para definir la previa — usar valores "razonables sin justificación" es proyectar incertidumbre como si fuera conocimiento.
- Se está comparando entre poblaciones — usar la misma previa para todas puede borrar diferencias reales.
En estos casos, conviene usar previas débiles (poco peso) o reportar resultados con y sin previa para mostrar la sensibilidad a la elección.
:::
**Recomendación general:** asigna a las previas un peso menor (preferiblemente mucho menor) que el tamaño de muestra observado. Esto garantiza que los datos reales tengan mayor peso en el análisis y evita que el conocimiento previo domine injustamente los resultados.
**Resultado**
La nueva matriz de transición:
- Es ergódica (todas las etapas están conectadas directa o indirectamente).
- Es reducible, lo que significa que algunas etapas pueden no ser alcanzables desde otras, dependiendo del ciclo de vida.
- Presenta tasas de mortandad y transición más realistas, gracias a la incorporación del conocimiento experto.
------------------------------------------------------------------------
## Intervalos de credibilidad para las transiciones
Una vez ajustada la matriz de transición con los datos observados y las previas informativas, podemos calcular los intervalos de credibilidad (ICr) para cada entrada de la matriz.
::: callout-note
**Por qué Dirichlet (transiciones) y Gamma (fecundidad).** Cada columna de una matriz de transición es un vector de probabilidades que suman 1. La distribución natural sobre tales vectores es la **distribución Dirichlet**, generalización multidimensional de la Beta. Al observar transiciones (datos multinomiales) y combinar con una previa Dirichlet, la posterior **también es Dirichlet** (es una previa conjugada) — esto hace los cálculos exactos y rápidos. Para cada entrada individual de la matriz, la distribución marginal es **Beta**, lo cual permite calcular intervalos de credibilidad directamente con `qbeta()`. La fecundidad, por ser un conteo positivo no acotado, se modela con una previa **Gamma** sobre la tasa de Poisson.
:::
::: callout-note
**Intervalo de confianza (IC) versus intervalo de credibilidad (ICr).** Aunque suenan parecidos, significan cosas distintas:
- **IC frecuentista al 95%:** "si repitiéramos este estudio infinitas veces, el 95% de los intervalos calculados contendrían el valor verdadero". No es una afirmación sobre el parámetro en este estudio.
- **ICr bayesiano al 95%:** "dada la previa y los datos, hay un 95% de probabilidad de que el verdadero valor del parámetro esté en este intervalo". Es una afirmación directa sobre el parámetro.
Lo que la mayoría de las personas *intuitivamente quieren decir* al leer un IC frecuentista es la interpretación bayesiana — y los intervalos de credibilidad son los que efectivamente se la dan.
:::
**¿Cómo se calculan?** Cada elemento de la matriz de transición ajustada sigue una distribución beta, ya que proviene de una distribución posterior marginal de una Dirichlet (multinomial-Dirichlet). Esto permite calcular intervalos de credibilidad para las tasas de transición.
Para obtener estos valores, se puede usar el tipo de retorno TN, que proporciona:
- La estimación puntual.
- El límite inferior del intervalo de credibilidad al 95%.
- El límite superior del intervalo de credibilidad al 95%.
### Ejemplo: etapa de plántula
Supongamos que estamos analizando las transiciones desde la etapa de plántula (s). Si el número de transiciones observadas es 2, y el peso de las previas es 1, el tamaño de muestra efectivo es 3. Esto genera un intervalo de credibilidad más amplio, reflejando la alta incertidumbre en la estimación. Por ejemplo:
- La tasa de mortandad puntual es 0.31.
- El intervalo de credibilidad al 95% va de 0.096 a 0.585.
Este rango amplio indica que el mejor estimado es muy incierto, lo cual es típico cuando se trabaja con tamaños de muestra pequeños.
### Interpretación
Un intervalo de credibilidad amplio significa que hay mucha incertidumbre en la estimación del parámetro. Esto resalta la importancia de contar con:
- Más datos de campo.
- Previas bien fundamentadas.
- Un análisis cuidadoso del tamaño de muestra efectivo.
```{r bayes15}
TN <- fill_transitions(
TF,
N,
P = RLT_Tprior,
priorweight = 0.1,
returnType = "TN"
)
a <- TN[, 1] # cambie 1 a 2, 3 etc para obtener la distribución beta marginal de cada columna (etapa de vida).
b <- sum(TN[, 1]) - TN[, 1] # cambie 1 a 2, 3 etc para obtener la distribución beta marginal de cada columna.
p <- a / (a + b)
lcl <- qbeta(0.025, a, b) # el valor 0.025 de la distribución beta
ucl <- qbeta(0.975, a, b) # el valor 0.975 de la distribución beta
knitr::kable(sprintf("%.4f (%.4f, %.4f)", p, lcl, ucl)) # Crear una tabla con los resultados con 4 cifras decimales (note la última fila son los fallecidos)
```
Una de las grandes ventajas de usar el paquete `raretrans` es que podemos calcular los intervalos de credibilidad de las tasas de transición y fecundidad de manera rápida y sencilla. Además, los intervalos de credibilidad están limitados a los valores entre 0 y 1, lo que es importante para las tasas de transición y fecundidad. Esto es especialmente útil cuando se trabaja con datos de campo donde el tamaño de muestra es pequeño y las transiciones raras son comunes. Muchos métodos estadísticos no pueden calcular intervalos de confianza para tasas de transición y fecundidad, y cuando lo hacen tienen valores ilógicos, como proporciones negativas o mayores a 1.
------------------------------------------------------------------------
### El efecto del tamaño de muestra sobre los intervalos de credibilidad
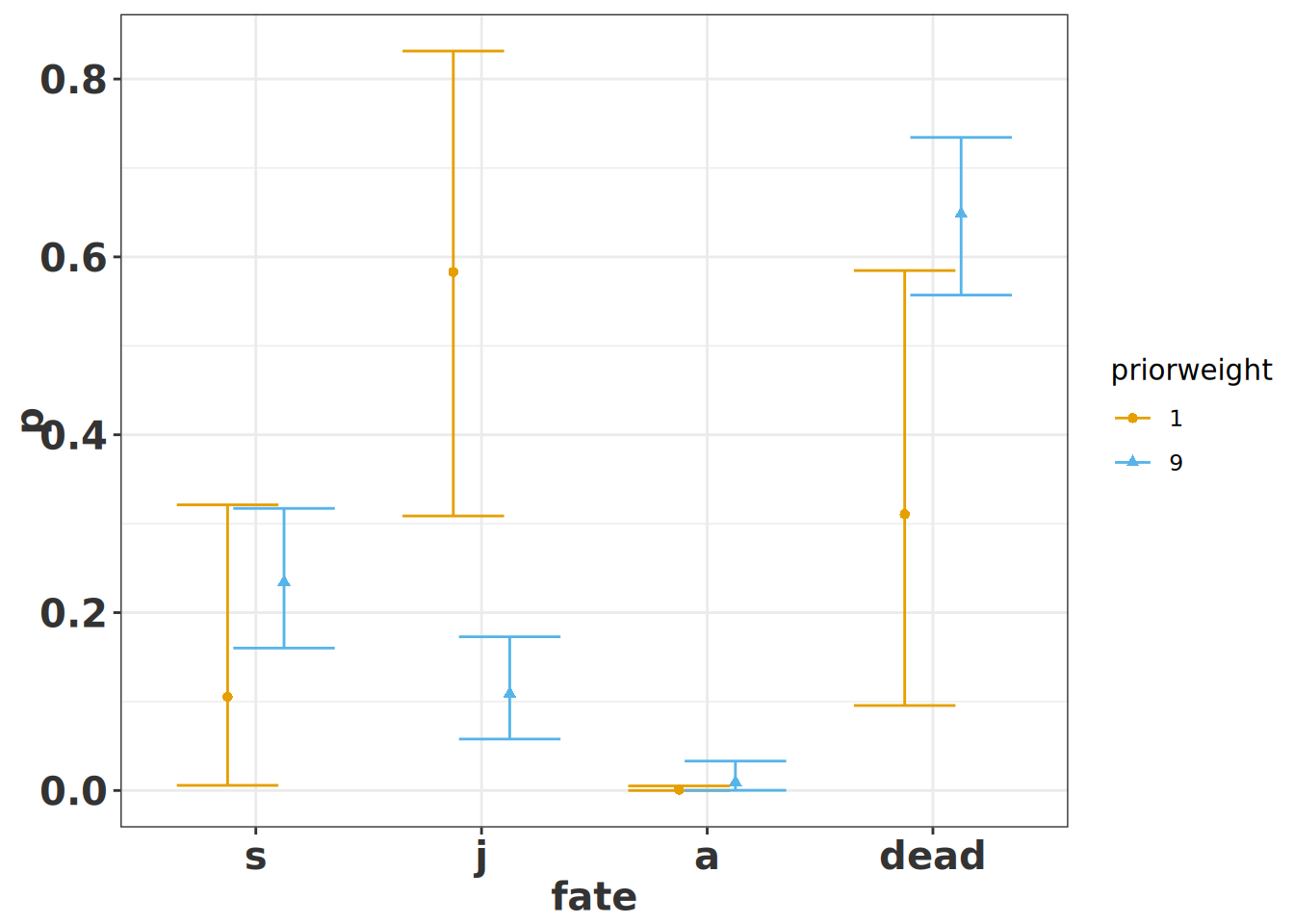
Cuando se aumenta el **tamaño de muestra efectivo**, los **intervalos de credibilidad** se vuelven más estrechos, lo que indica mayor confianza en las estimaciones puntuales. Aumentar el tamaño de muestra efectivo a $20$ especificando: `priorweight`$= 9 (9*2 = 18 + 2 = 20)$ hace que los intervalos de credibilidad se reduzcan bastante. Entonces, es importante enfatizar en que el tamaño de muestra tiene gran impacto sobre la confianza que se tiene sobre el estimador puntual (el promedio) de las transiciones, permanencias y mortalidad. Note en la figura como el tamaño del intervalo de credibilidad cambia drásticamente cuando se aumenta el número de datos.
El intervalo de credibilidad se estrecha cuando el tamaño de muestra efectivo aumenta, reflejando mayor confianza en el estimador puntual.
IMPORTANTE: Aunque este ejemplo muestra cómo el tamaño de muestra afecta la credibilidad, en la práctica:
- El peso de las previas (priorweight) debe ser menor que el tamaño de muestra observado.
- Esto asegura que los datos reales del campo tengan mayor influencia en el análisis.
- Usar un tamaño de muestra a priori demasiado grande puede sesgar los resultados y reducir la validez del modelo.
```{r}
#| label: fig-bayes16
#| fig-cap: "Probabilidades posteriores de transición por destino para dos pesos del prior (1 y 9), representadas con puntos y barras de error del intervalo de credibilidad del 95%."
TN <- fill_transitions(
TF,
N,
P = RLT_Tprior,
priorweight = 9,
returnType = "TN"
)
a <- TN[, 1]
b <- sum(TN[, 1]) - TN[, 1] # El parámetro beta es la suma de todos los demás parámetros de Dirichlet.
p2 <- a / (a + b)
lcl2 <- qbeta(0.025, a, b)
ucl2 <- qbeta(0.975, a, b)
alltogethernow <- tibble(
priorweight = factor(rep(c(1, 9), each = 4)),
fate = factor(
rep(c("s", "j", "a", "dead"), times = 2),
levels = c("s", "j", "a", "dead")
),
p = c(p, p2),
lcl = c(lcl, lcl2),
ucl = c(ucl, ucl2)
)
ggplot(
data = alltogethernow,
mapping = aes(x = fate, y = p, group = priorweight, colour = priorweight)
) +
geom_point(
mapping = aes(shape = priorweight),
position = position_dodge(width = 0.5)
) +
geom_errorbar(
mapping = aes(ymin = lcl, ymax = ucl),
position = position_dodge(width = 0.5)
) +
theme_bw() +
rlt_style_colour()
```
------------------------------------------------------------------------
## Intervalo de credibilidad para la tasa de crecimiento asintótica $\lambda$
Obtener intervalos de credibilidad para la tasa de crecimiento asintótica, $\lambda$, requiere simular matrices a partir de las distribuciones posteriores. Estas simulaciones usualmente son complicadas de hacer, por lo que hemos incorporado en el paquete la función `raretrans::sim_transitions()` para generar una lista de matrices simuladas dada la matriz observada y las previas. En otras palabras, esta función calcula múltiples veces las matrices de transición y fecundidad basándose en los datos observados y en los valores *a priori* especificados, pero variando los valores de los parámetros de las distribuciones beta y gamma.
### Simulación de una matriz de transición
Siguiendo el marco bayesiano que hemos venido trabajando en este capítulo, es necesario especificar las previas de la fecundidad como una matriz. Las celdas de la matriz de los estados en los que no hay reproducción deben ser `NA`, usando `NA_real_`, el cual es un valor que no está presente pero podría aceptar valores con puntos decimales. Nota que el valor *a priori* de la fecundidad es 0.0001.
```{r bayes17}
alpha <- matrix(c(
NA_real_, NA_real_, 1e-4,
NA_real_, NA_real_, NA_real_,
NA_real_, NA_real_, NA_real_
), nrow = 3, ncol = 3, byrow = TRUE)
beta <- matrix(c(
NA_real_, NA_real_, 1e-4,
NA_real_, NA_real_, NA_real_,
NA_real_, NA_real_, NA_real_
), nrow = 3, ncol = 3, byrow = TRUE)
fill_fertility(TF, N, alpha = alpha, beta = beta)
```
El cambio en la fecundidad es \< 0.0001 en comparación con el valor observado.
### Una simulación de la matriz de transición
Corra el script múltiples veces, y verá que los valores cambian en cada simulación. En este script la simulación es solamente una vez (`samples = 1`). Este script es para que visualicen lo que hace la función `sim_transitions()`.
```{r bayes18}
sim_transitions(
TF,
N,
P = RLT_Tprior,
alpha = alpha,
beta = beta,
priorweight = 0.5,
samples = 1
)
```
### Simulación de 5000 matrices de transición
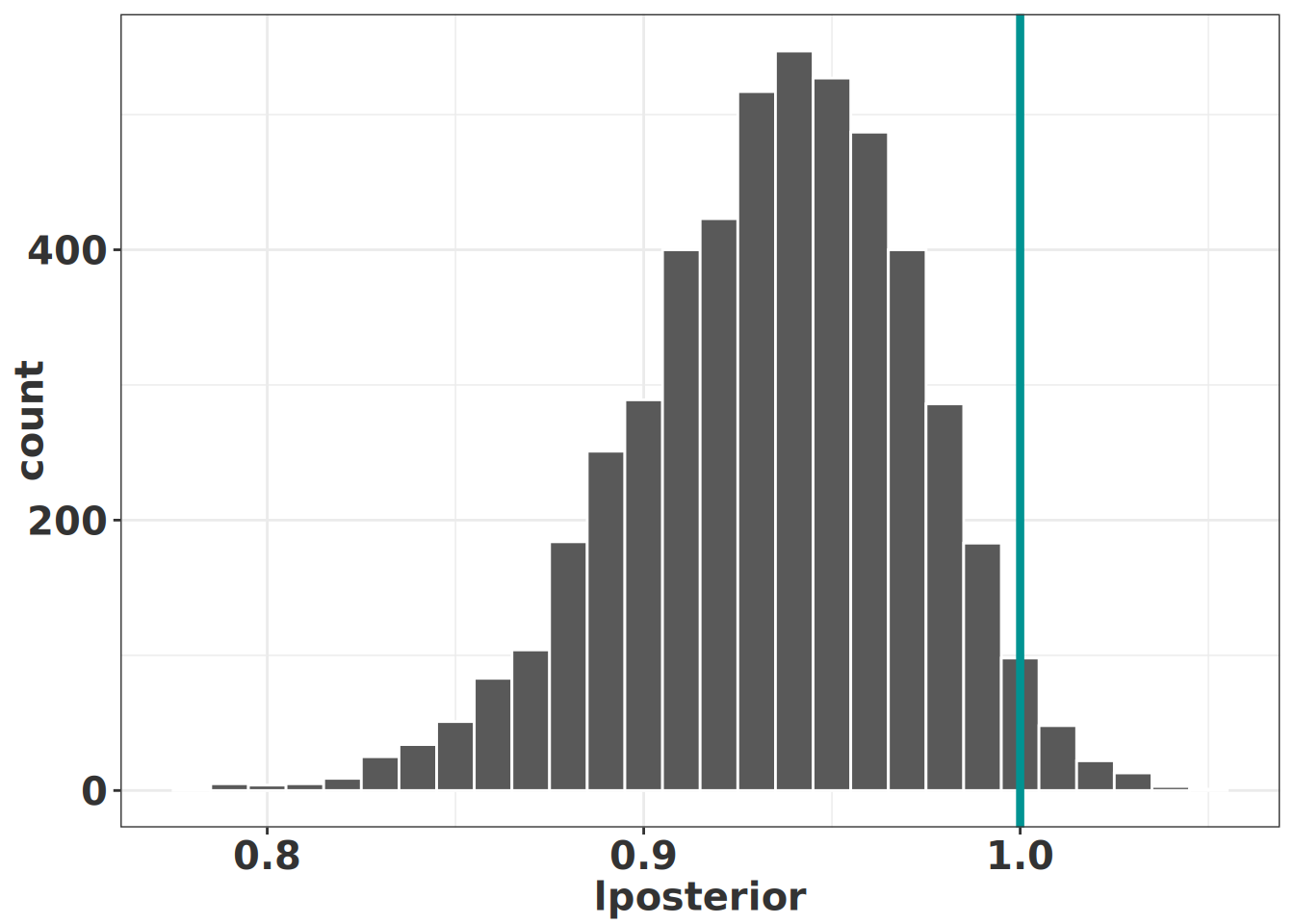
Ahora simulamos 5000 veces, calculamos el valor $\lambda$ de cada matriz y creamos un histograma de su distribución. Se añaden los valores de lambda en un objeto llamado `RLT_0.5`. Se visualiza la distribución de $\lambda$ usando un histograma. Note que el valor de $\lambda = 1$ queda dentro de la distribución; en otras palabras, el histograma muestra que la mayoría de los valores de $\lambda$ están cerca de 1.
```{r}
#| label: fig-bayes19
#| fig-cap: "Histograma de la distribución posterior de la tasa finita de crecimiento λ obtenida a partir de 5000 matrices simuladas, con una línea vertical en λ = 1."
set.seed(8390278) # make this part reproducible
alpha2 <- matrix(c(
NA_real_, NA_real_, 0.025,
NA_real_, NA_real_, NA_real_,
NA_real_, NA_real_, NA_real_
), nrow = 3, ncol = 3, byrow = TRUE)
beta2 <- matrix(c(
NA_real_, NA_real_, 1,
NA_real_, NA_real_, NA_real_,
NA_real_, NA_real_, NA_real_
), nrow = 3, ncol = 3, byrow = TRUE)
# generar 5000 matrices basadas en las previas de transiciones y de fecundidades, el tamaño de muestra, además de los datos
RLT_0.5s <- sim_transitions(TF, N,
P = RLT_Tprior, alpha = alpha2, beta = beta2,
priorweight = 0.5, samples = 5000
)
# extraer los valores de lambda de cada simulación en un histograma
RLT_0.5 <- tibble(lposterior = map_dbl(RLT_0.5s, lambda)) # convertir la lista en un tibble
ggplot(
data = RLT_0.5,
mapping = aes(x = lposterior)
) +
geom_histogram(binwidth = 0.01, colour = "white") +
theme_bw() +
rlt_style_fill() +
geom_vline(
xintercept = 1,
color = "#009392", linewidth = 1.5
)
```
------------------------------------------------------------------------
## Probabilidad posterior de que la población esté creciendo (λ \> 1)
A diferencia de un contraste de hipótesis frecuentista (donde se obtiene un valor-p), el marco bayesiano nos permite responder directamente la pregunta de interés: **¿con qué probabilidad la población está creciendo?** Lo hacemos contando qué proporción de las simulaciones de la distribución posterior produjo $\lambda > 1$. En este caso, $P(\lambda > 1 \mid \text{datos}, \text{previas}) = 0.14$, lo que indica que sólo en el 14% de los escenarios consistentes con los datos y la previa la población crece. La evidencia para crecimiento poblacional es débil; lo más probable es que la población esté en declive. La estadística `p_increase` reporta exactamente este valor.
```{r bayes20}
RLT_0.5_summary <- summarize(
RLT_0.5,
medianaL = median(lposterior),
promedioL = mean(lposterior),
skewnessL = e1071::skewness(lposterior, type = 2),
ICrBajo = quantile(lposterior, probs = 0.025),
ICrAlto = quantile(lposterior, probs = 0.975),
p_increase = sum(lposterior > 1.) / n()
)
```
### Tabla de resultado de lambda
```{r bayes21}
flextable(signif(RLT_0.5_summary, digits = 2))
```
------------------------------------------------------------------------
## ADDENDUM
::: callout-important
**Nunca uses previas uniformes en análisis poblacionales reales.** Una previa uniforme (e.g., Dirichlet con todos los parámetros iguales) asigna la misma probabilidad a transiciones biológicamente plausibles y a transiciones imposibles (un adulto reproductivo "regresando" a plántula, una ballena bebé pasando directamente a madre, una semilla transformándose en árbol adulto en un periodo). El resultado es una matriz matemáticamente válida pero **biológicamente sin sentido**, que infla la incertidumbre con escenarios irreales y puede ocultar señales legítimas en los datos.
**Siempre construye previas informadas** que reflejen el ciclo de vida de la especie: marca con 0 las transiciones imposibles, asigna probabilidades pequeñas a las raras pero posibles, y concentra masa en las transiciones que la biología espera. La regla práctica es: si no puedes defender la previa frente a un especialista de la especie, no la uses.
La sección que sigue muestra qué pasa con una previa uniforme **únicamente con fines didácticos** — para que se vea de primera mano por qué falla. No la copies en un análisis de tu propia especie.
:::
### Uso de previas uniformes
Esta sección es para demostrar por qué no es recomendable el uso de distribuciones previas uniformes. Si se entiende el concepto de valores *a priori* y *a posteriori*, no es necesario leer esta sección.
### Matriz de transición
A continuación vamos a añadir una distribución Dirichlet uniforme como previa con un peso = $1$ a la matriz de transición, $T$. En este caso, tenemos 4 destinos (3 + muerte), por lo que cada destino añade 0.25 a la matriz de destinos *observados* (¡no a la matriz de transición!). Cuando especificamos una matriz con valores *a priori* para las transiciones, hay una fila más que columnas. Esta fila extra representa la mortalidad por etapa.
Note que no se recomienda el uso de una previa uniforme; aquí se utiliza sólo para mostrar el concepto. Una distribución previa uniforme no refleja un ciclo de vida razonable para especies biológicas. Por ejemplo, una previa uniforme podría hacer que las plántulas pasen a adulto de un tiempo a otro o que los adultos regresen a ser plántulas, lo cual no tiene mucho sentido biológico en muchas especies. Considera lo siguiente: ¿es posible que una ballena bebé pueda pasar a ser madre al año siguiente? Añadir una transición de 0.25 de ballena bebé a madre no tiene sentido biológico.
Note que ahora la matriz de transición es ergódica pero no tiene sentido biológico. Compare la matriz con las previas uniformes versus la matriz original.
```{r bayes22}
Tprior <- matrix(0.25, byrow = TRUE, ncol = 3, nrow = 4) # crear la matriz de previas uniformes
mat_to_ft(Tprior)
Posterior_con_Unif <- fill_transitions(TF, N, P = Tprior) # resultado de la matriz de transición básica con las previas
mat_to_ft(Posterior_con_Unif)
# Para entender las diferencias del impacto compara con la matriz de transición
# original `$T` del objeto `TF` (popbio::projection.matrix con TF=TRUE devuelve
# una lista con $T y $F).
mat_to_ft(TF$T)
mat_to_ft(TF$F)
```
------------------------------------------------------------------------
### ¿Cómo calcular en forma directa las transiciones de estados?
Podemos obtener el resultado haciendo los cálculos directamente. Lo primero que necesitamos es el vector de observaciones porque la distribución posterior se calcula a partir de las observaciones de transiciones, no de la matriz de transiciones. Si no tiene experiencia con análisis Bayesiano es recomendable seguir los siguientes scripts
```{r bayes23}
Tobs <- sweep(TF$T, 2, N, "*") # obtener las observaciones de transiciones
Tobs <- rbind(Tobs, N - colSums(Tobs)) # añadir la fila de muerte
Tobs <- Tobs + 0.25 # añadir los valores de las previas
sweep(Tobs, 2, colSums(Tobs), "/")[-4, ] # dividir por la suma de la columna y descarta la fila de muerte
```
La previa uniforme "rellena" las transiciones faltantes, pero también crea problemas porque algunos de los valores de transición que se asignan son biológicamente imposibles en muchas especies. Por ejemplo, para la transición de adulto a plántula se asigna un valor; sin embargo, este valor sólo es posible en la matriz de fecundidad $F$ en las especies de *Lepanthes*. Debido a esto, no es recomendable el uso de previas uniformes, ya que no toman en cuenta el ciclo de vida de nuestra especie. Será necesario crear una matriz de *priors* que refleje el ciclo de vida de la especie de interés.
------------------------------------------------------------------------