---
bibliography: book.bib # Bibliografía para tablas tibble
description: "Cómo datos sin sentido biológico afectan los modelos matriciales de proyección poblacional: ejemplos, diagnósticos y recomendaciones para evitar errores frecuentes."
image: "images/Erycina_crista-galli_Diana_Molina_Ozuma.jpg"
open-graph:
description: "Cómo datos sin sentido biológico afectan los modelos matriciales de proyección poblacional: ejemplos, diagnósticos y recomendaciones para evitar errores frecuentes."
twitter-card:
description: "Cómo datos sin sentido biológico afectan los modelos matriciales de proyección poblacional: ejemplos, diagnósticos y recomendaciones para evitar errores frecuentes."
---
# Impactos de datos sin sentido biológico {#ImpactosSinSentido}
Por: Raymond L. Tremblay
#### Librerías de R requeridas para el siguiente módulo
```{r, message=FALSE, warning=FALSE}
#| docx_fit: true
#| label: fig-sin_sentido_1
#| fig-cap: "Bloque de configuración que instala y carga los paquetes DiagrammeR, Rage, popdemo, popbio, MCMCpack y otros, junto con las funciones auxiliares de figuras."
# devtools::install_github("rich-iannone/DiagrammeR") # Instalar directamente de github para tener la versión más reciente
if (!require("pacman")) {
install.packages("pacman")
}
pacman::p_load(
"DiagrammeR",
"Rage",
"popdemo",
"popbio",
"interpretCI",
"MCMCpack",
"ggplot2",
"plyr",
"reshape2"
)
library(DiagrammeR)
library(Rage)
library(popdemo)
library(popbio)
library(tidyverse)
library(flextable)
library(interpretCI)
library(MCMCpack)
library(plyr)
library(reshape2)
source("R/figuras_helpers.R") # plc_save(), grviz_save(), save_plot_png()
```
```{r ssin-theme}
rlt_theme <- theme(
text = element_text(family = "Libertinus Serif"),
axis.title.y = element_text(colour = "grey20", size = 10, face = "bold"),
axis.text.x = element_text(colour = "grey20", size = 10, face = "bold"),
axis.text.y = element_text(colour = "grey20", size = 15, face = "bold"),
axis.title.x = element_text(colour = "grey20", size = 15, face = "bold")
) +
theme(
# Remover los bordes
panel.border = element_blank(),
# Remover las líneas
panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
# Remover el fondo del panel
panel.background = element_blank(),
# añadir lineas más gruesas
axis.line.x = element_line(colour = "black", linewidth = 1),
axis.line.y = element_line(colour = "black", linewidth = 1)
)
diverging <- c("#E69F00", "#56B4E9", "#009392", "#F0E442", "#0072B2", "#D55E00", "#CC79A7", "#000000")
# --- 2. Combinarlos en un objeto de tipo lista ---
# Esta lista se puede añadir al gráfico con un solo '+'
rlt_style_fill <- function() {
list(
rlt_theme,
scale_fill_manual(values = diverging)
)
}
rlt_style_colour <- function() {
list(
rlt_theme,
scale_colour_manual(values = diverging)
)
}
```
El valor principal de los estudios científicos radica en el supuesto de que los datos obtenidos representan una versión suficientemente cercana de la realidad biológica de campo, de modo que puedan utilizarse para inferir sobre la ecología y conservación de las especies de interés. Este supuesto de que los datos representan realmente a la realidad es un mito, ya que solamente son una muestra, que esperamos que se acerque lo suficiente para tener confianza en los resultados. En ningún estudio ecológico se podrán obtener **TODOS** los datos para evaluar todas las relaciones bióticas y abióticas que mantiene un organismo y sus interacciones. Dicho modelo sería demasiado complejo e irreal dentro del concepto de lo que se puede lograr en un trabajo científico. Es por ello que, el objetivo de estos trabajos es tener una apreciación de los parámetros más importantes para definir patrones e interacciones suficientemente cercanas a la realidad para ser útiles. Por consiguiente, la base de estos estudios está sostenida en las unidades de muestreo y en la recolección de datos. Si los datos no representan la realidad, las interpretaciones de los análisis pueden ser erróneas. La matemática, con sus ecuaciones y sus modelos, no hará que la biología se aproxime más a la realidad, si desde el principio los datos y el método de recolección de datos son erróneos.
Es por ello que en esta sección evaluaremos diferentes aspectos de los análisis matriciales poblacionales de proyección (MPP) y diferentes aspectos de la recolección de datos que pudiesen ser problemáticos cuando uno considera la biología de una especie. La lista de *Impactos* no pretende incluir todos los posibles efectos de datos sin sentido; sino más bien, brindar algunos ejemplos de lo que pasa cuando uno no considera estos problemas y cómo pueden distorsionar las interpretaciones. Por lo tanto, se les extiende una advertencia a todos los biólogos de poblaciones, para que estén conscientes de estos temas y siempre evalúen críticamente sus datos desde la forma de recolección, su análisis e interpretaciones de sus resultados y aceptar que son solamente un modelo (una caricatura) de muchos posibles modelos.
------------------------------------------------------------------------
## Cuatro familias de errores
Los problemas que pueden invalidar un análisis matricial poblacional caen en cuatro grandes grupos. Conocerlos por separado ayuda a saber **dónde mirar** cuando algo no cuadra:
1. **Errores en la recolección de datos.** Tamaños de muestra pequeños, eventos raros que nunca se observaron en el periodo de estudio, etapas crípticas (semillas, latencia, juveniles ocultos) que no se detectan. Síntoma típico: probabilidades de transición exactamente iguales a 0 o a 1.
2. **Errores en la construcción de la matriz.** Supervivencia perfecta (columnas que suman 1 sin reproducción), columnas que suman *más* de 1 por redondeo excesivo, transiciones biológicamente posibles que faltan en la matriz (matrices reducibles).
3. **Errores en la fecundidad.** Usar el número crudo de semillas/huevos en vez de los individuos que efectivamente reclutan al primer estadio del modelo, omitir el banco de semillas, no especificar si el censo es pre- o post-reproductivo (ver el [protocolo estandarizado del capítulo 121](#protocolo)).
4. **Errores en la inferencia estadística.** Calcular intervalos de confianza con la distribución normal sobre proporciones acotadas en \[0, 1\] —que produce límites negativos o mayores que 1—, no comunicar la incertidumbre de los parámetros.
Para cada error mostraremos tres cosas: **cómo se ve** en el código y la matriz, **cómo detectarlo en R**, y **cómo evitarlo o corregirlo**. Así el capítulo sirve tanto como guía de lectura crítica de la literatura como lista de verificación al construir su propio MPP.
::: callout-tip
## ¿Cuándo regresar a este capítulo?
Este capítulo está diseñado para consultarse en dos momentos distintos: **antes** de comenzar el muestreo, para anticipar qué decisiones de diseño afectarán la matriz, y **después** de tener una matriz preliminar, para diagnosticarla antes de pasar a inferencias o publicación. Léalo con su matriz al lado.
:::
------------------------------------------------------------------------
## Tamaños de muestra pequeños o eventos raros
Sin duda, el tema principal para el uso de MPP ha sido evaluar la proyección de población de especies raras o en peligro de extinción [@caswell2000matrix; @gascoigne2023standard]. En consecuencia, las especies raras o en peligro de extinción son naturalmente pequeñas y el tamaño de la muestra del estudio o de algunas de las etapas/edades del ciclo de vida son frecuentemente pequeños. Estos tamaños de muestra reducidos suelen provocar estimaciones sesgadas de los parámetros y, a menudo, resultados sin sentido biológico. Algunos de los problemas incluyen no haber observado eventos raros (transiciones poco comunes), como la muerte de un adulto o la germinación de una semilla. En estos casos, la probabilidad de supervivencia o transición pudiese ser de cero, lo que pudiese resultar en una matriz reducible o no irreducible.
------------------------------------------------------------------------
### Supervivencia perfecta
Muestreos en donde no se observa mortalidad, que equivaldrían a una supervivencia perfecta, son problemáticos. Esto se pudiese observar generalmente en especies longevas, en donde la mortalidad de los individuos, en especial de los individuos adultos es rara, pero la probabilidad existe. Considere una especie donde se recopilan datos de la especie **Sp1** y se crea una matriz de transición **Sp1matU** y una matriz de fertilidad **Sp1matF**. Note que ninguno de los individuos adultos muestreados murió, por lo que el valor de la permanencia es de 1 (supervivencia perfecta). En consecuencia, el tamaño de la población nunca disminuye. Sin embargo, esto es completamente irreal. Así, bajo todos los modelos biológicamente realistas, esperaríamos que el tamaño de la población comience a disminuir si una población no tiene reproducción. Sin embargo, en nuestro ejemplo, a pesar de que la fecundidad es cero, nuestra población se mantiene ya que los individuos adultos presentan una supervivencia perfecta, lo cual es biológicamente imposible.
```{r, message=FALSE}
#| docx_fit: true
#| label: fig-sin_sentido_2
#| fig-cap: "Ciclo de vida correspondiente a la matriz Sp1matA con estadios plántula, juvenil y adulto, sin fecundidad y lambda igual a 1."
Sp1matU <- rbind(
c(0.0, 0.0, 0.0),
c(0.5, 0.3, 0.0),
c(0.0, 0.4, 1.0)
) # transition matrix
Sp1matF <- rbind(
c(0.0, 0.0, 0.0),
c(0.0, 0.0, 0.0),
c(0.0, 0.0, 0.0)
) # fertility matrix
Sp1matA <- Sp1matU + Sp1matF # TF matrix
lambda(Sp1matA) # lambda es 1, lo que indica que la población no cambia
stages <- c("plantula", "juvenil", "adulto")
plc_save(Sp1matA, stages = stages, fontsize = 0, png = "images/sin_sentido_2_Sp1matA.png")
```
Si analizamos **Sp1matU**, asumimos que ninguno de los individuos muestreados en etapa adulta muere, en ningún momento!!! Claramente esto no es realista. Para cualquier especie, la probabilidad de muerte en cualquier etapa nunca es cero (aunque podría ser muy pequeña) y, por lo tanto, nuestra matriz no tiene sentido biológico. Considere una especie de árbol, como los árboles de *Sequoia*, en donde es muy probable que la supervivencia de los árboles grandes sea muy alta entre un año y otro y por lo tanto la **supervivencia observada** sea del 100%, pero, este 100% no es realista a largo plazo. Hay que diferenciar entre lo que se observa en un periodo de muestreo y las probabilidades a largo plazo. Es posible que en el sitio de estudio NO se observó mortalidad de los árboles grandes entre los dos periodos de muestreo, lo que resultaría en una mortalidad de cero, sin embargo, este valor no es real a largo plazo. Ese estimado de campo puede ser un resultado de un tamaño de muestra insuficiente o de eventos raros (aun con tamaños de muestra grandes).
En el siguiente *script* mostramos que la población no cambia después de 7-8 periodos/años y se mantiene cercana a uno (usamos la matriz de transición sin fecundidad). Si no hay reclutas (matriz de fertilidad), el tamaño de la población debería reducirse con el tiempo. Todos los modelos de transiciones que no incluyan a la matriz de fertilidad (solamente la matU), deberían resultar en reducción de tamaño poblacional en el tiempo, debido a la no incorporación de nuevos individuos (fecundidad), de la pérdida de individuos muestreados (mortalidad).
```{r sin_sentido_3}
n <- c(50, 50, 50)
truelambda(Sp1matU) # después de unos periodos de tiempo vemos que el crecimiento poblacional es 1
pop.projection(Sp1matU, n = n)$pop.changes # lista del cambio poblacional en 19 años
```
::: callout-warning
## Cómo detectar y resolver — supervivencia perfecta
**Detección en R:**
``` r
any(diag(matU) >= 1) # ¿hay estasis con probabilidad 1?
any(colSums(matU) >= 1 & colSums(matF) == 0) # ¿columna que suma 1 sin reproducción?
```
**Causa típica:** tamaño de muestra insuficiente para observar la mortalidad rara de los adultos durante el periodo de estudio.
**Cómo evitarlo o corregirlo:**
1. Reportar el tamaño de muestra de cada etapa y la incertidumbre asociada (ver capítulo bayesiano).
2. Usar un previo bayesiano informado vía `raretrans` o una distribución de Dirichlet para inferir una mortalidad pequeña pero `> 0`.
3. Si se decide mantener `0.995` o similar, justificar el valor con literatura sobre la longevidad de la especie y reconocer la limitación.
:::
------------------------------------------------------------------------
#### Un cambio pequeño en la mortalidad.
El efecto de aumentar el tamaño de muestra impacta directamente los posibles valores en la matriz. Considere la siguiente situación. Si tenemos solamente cinco individuos en la etapa uno, ¿cuáles son los posibles valores de mortalidad? Esto dependería del número de individuos que mueran, que pudiera ser ninguno, 1, 2,... o todos. Entonces las probabilidades que pudieran entrar en la matriz dependiendo el número de individuos muertos serían: 0, 0.2, 0.4, 0.6, 0.8, 1.0. Note que los valores intermedios no pueden existir en la matriz. Por lo tanto, si uno tiene un tamaño de muestra pequeño, es probable que los valores en la matriz no representen la realidad biológica, pero si representan lo observado en el muestreo de campo.
Si consideramos la matriz del script 3 y observamos el valor de la permanencia (sobrevivencia) de los individuos adultos podemos deducir que se muestrearon 1000 individuos y que 995 sobrevivieron. Así con tamaños muestrales superiores, por ejemplo 1000, además de conferir una mayor confianza en el parámetro, permite la existencia de valores intermedios. Tenga en cuenta que en este nuevo modelo de la especie *SP1*, aunque la tasa de supervivencia es muy cercana a 100% (0.995), existe mortalidad y por tanto existiría una disminución de la población en cada período de tiempo (aunque solo sea en una pequeña fracción), si los valores de fecundidad fueran de cero. Siempre hay que considerar si esta tasa de mortalidad es biológicamente real o es un resultado del tamaño de la muestra.
```{r sin_sentido_4}
Sp1matU_2 <- rbind(
c(0.0, 0.0, 0.0),
c(0.5, 0.3, 0.0),
c(0.0, 0.4, 0.995)
) # transition matrix
Sp1matF <- rbind(
c(0.0, 0.0, 1.0),
c(0.0, 0.0, 0.0),
c(0.0, 0.0, 0.0)
) # fertility matrix
Sp1matA_2 <- Sp1matU_2 + Sp1matF
lambda(Sp1matA_2) # lambda es 0.995, lo que indica que la población disminuye
pop.projection(Sp1matU_2, n = n)$pop.changes # lista del cambio poblacional en 19 años
```
::: img-float
{#fig-erycina-crista-galli-diana-molina-ozuma style="float: left; margin: 10px;width: 40% ; "}
:::
En la mayoría de las investigaciones el tamaño de muestra es limitado. Por lo tanto, es importante considerar si la tasa/transición de mortalidad es realista y si el tamaño de la muestra es suficiente para estimar la tasa/transición de mortalidad con confianza. Si la tasa de mortalidad es muy baja, cercana a cero o a 100%, es probable que el tamaño de la muestra sea demasiado pequeño para estimar la tasa de mortalidad con precisión. En consecuencia, la tasa de mortalidad estimada puede estar lejos de la realidad si hubiese tenido un estimado basado en un *N* más grande.
------------------------------------------------------------------------
<br>
<br>
<br>
<br>
## ¿Cuáles son los tamaños de muestra típicos en los estudios de orquídeas?
Usando el artículo de Ticktin y colaboradores [@ticktin2020synthesis], vemos algunos de los tamaños de muestra de los estudios de orquídeas usando MPP. Esta tabla representa solamente parte de los datos en la publicación. El objetivo es demostrar el tamaño de muestra en algunos de los estudios de orquídeas. Note que en algunos estudios el tamaño por etapa es muy pequeño, lo que puede resultar en estimaciones sesgadas de los parámetros y, a menudo, resultados sin sentido biológico.
```{r sin_sentido5, message=FALSE, echo=FALSE}
N_PPM <- tribble(
~Especie, ~"Población", ~Etapas, ~"Tamaño de Muestra", ~Referencia,
"*Erycina crista-galli*", "Pop_1", "Plántulas", 41, "@mondragon2007life",
"", "", "Juveniles", 50, "",
"", "", "Adultos 1", 221, "",
"", "", "Adultos 2", 226, "",
"*Erycina crista-galli*", "Pop_2", "Plántulas", 4, "@mondragon2007life",
"", "", "Juveniles", 32, "",
"", "", "Adultos 1", 25, "",
"", "", "Adultos 2", 531, "",
"*Euchile karwinskii*", "Sin cosecha", "Plántulas", 20, "@elliott2014demography",
"", "", "Juveniles 1", 74, "",
"", "", "Juveniles 2", 91, "",
"", "", "Adultos", 44, "",
"*Euchile karwinskii*", "Con cosecha", "Plántulas", 3, "@elliott2014demography",
"", "", "Juveniles 1", 26, "",
"", "", "Juveniles 2", 47, "",
"", "", "Adultos", 20, "",
"*Lepanthes caritensis*", "Carite", "Plántulas", 4, "@tremblay1997lepanthes",
"", "", "Juveniles", 54, "",
"", "", "Adulto no reproductivo", 24, "",
"", "", "Adultos", 12, "",
"*Oncidium poikilostalix*", "Sin cosecha", "Etapa 1", 284, "@garcia2017impact",
"", "", "Etapa 2", 194, "",
"", "", "Etapa 3", 270, "",
"", "", "Etapa 4", 338, ""
)
list_fl <- N_PPM %>%
flextable() %>%
ftExtra::colformat_md(j = c("Especie", "Referencia")) # *italics* de especie + resuelve @citas de la columna Referencia
list_fl
```
------------------------------------------------------------------------
### Irreducibilidad: cuando faltan transiciones entre etapas
La *irreducibilidad* es un concepto importante en los análisis de matrices poblacionales. Como lo muestra [@caswell2000matrix] y más recientemente por [@stott2010reducibility], las matrices deben de ser *irreducibles*. El concepto de *irreducibilidad* está asociado con el ciclo de vida de la especie y la matriz debe incluir las transiciones de todas las etapas a todas las demás etapas. En la figura del ciclo de vida siguiente, faltan dos componentes importantes en la historia de vida de la especie, ningún juvenil crece para convertirse en adulto y ninguno de los adultos produce plántulas (semillas que crecen hasta convertirse en plántulas).
Por tal motivo, la matriz sugiere que los juveniles no crecen para convertirse en adultos, y todos los juveniles siguen siendo juveniles o mueren, ya que no hay una flecha que conecte a los juveniles con los adultos. En tanto que en el caso de los adultos no existe una flecha que los conecte con las plántulas, lo que sugiere que no hay producción de nuevas plántulas por parte de los adultos.
```{r}
#| docx_fit: true
#| label: fig-sin_sentido_6
#| fig-cap: "Ciclo de vida correspondiente a la matriz Sp1matA_NT con estadios plántula, juvenil y adulto, sin fecundidad y con retención del adulto de 0.995."
Sp1matU_NT <- rbind(
c(0.0, 0.0, 0.0),
c(0.5, 0.7, 0.0),
c(0.0, 0.0, 0.995)
) # transition matrix
Sp1matF <- rbind(
c(0.0, 0.0, 0.0),
c(0.0, 0.0, 0.0),
c(0.0, 0.0, 0.0)
) # fertility matrix
Sp1matA_NT <- Sp1matU_NT + Sp1matF # TF matrix
stages <- c("plantula", "juvenil", "adulto")
plc_save(Sp1matA_NT, stages = stages, fontsize = 0, png = "images/sin_sentido_6_Sp1matA_NT.png")
```
#### Función para evaluar si una matriz es irreducible
Una manera fácil de determinar si la matriz es irreducible es correr la función `isIrreducible()` del paquete `popdemo`. Tenga en cuenta que, en este caso, el resultado es *FALSE* para la matriz anterior. Para comparar, si evaluamos la matriz **Sp1matA_2** el resultado es **TRUE** ya que todas las etapas pueden llegar a todas las demás etapas. En este caso, la matriz es irreducible y por tanto cumple con los requisitos necesarios para muchos de los análisis de matrices poblacionales.
```{r sin_sentido_7}
isIrreducible(Sp1matU_NT) # para la matriz de transición, faltando una etapa de transición y fertilidad
isIrreducible(Sp1matA_NT) # para la matriz TF, falta una etapa de transición
# Ahora considerando Sp1matA_2
# En esta matriz que incluye todas las transiciones y la fertilidad el resultado es "TRUE"
Sp1matA_2
isIrreducible(Sp1matA_2)
```
::: callout-warning
## Cómo detectar y resolver — matriz reducible
**Detección en R:**
``` r
popdemo::isIrreducible(matA) # FALSE indica el problema
popdemo::isErgodic(matA) # complementaria: ¿la población converge?
```
**Causa típica:** una transición biológicamente posible (juvenil → adulto, adulto → plántula vía reproducción) tiene valor 0 en la matriz, ya sea por no haberse observado en el muestreo o por no haber incluido la fecundidad correctamente en la matriz F.
**Cómo evitarlo o corregirlo:**
1. Dibujar el gráfico del ciclo de vida y verificar que cada etapa puede llegar a cualquier otra etapa por algún camino.
2. Si la transición existe biológicamente pero es rara, asignarle un valor pequeño positivo (p. ej., `1/(N+1)`) en lugar de cero, o usar un previo bayesiano.
3. Si la transición no existe en la biología (etapas post-reproductivas, dispersión unidireccional), reportar la matriz como reducible *justificadamente* y elegir métodos compatibles con esta estructura (ver capítulo de Propiedades de las matrices).
:::
------------------------------------------------------------------------
### Ninguna supervivencia
La mortalidad es uno de los estadios del ciclo de vida de cada especie, pero raramente se añade al diagrama del ciclo de vida, porque es implícito en los cálculos ya que si restamos 1 a la suma de la columna de transición de un estadio dado, el resultado será la mortalidad de dicho estadio. A menudo se observa que la supervivencia de los individuos más pequeños o la primera etapa del ciclo de vida de una especie es muy reducida, por lo que la probabilidad de supervivencia es muy baja. Por ejemplo, la mayoría de las semillas no sobreviven para germinar. Esta es la norma en las orquídeas donde la producción de semillas es muy alta, a veces millones de semillas en una cápsula [@arditti2000numerical], pero los porcentajes de germinación en el campo suelen ser menores. Al momento no conocemos trabajos sistemáticos evaluando la probabilidad de germinación en orquídeas. Sin embargo, esto no se limita a las orquídeas, en los árboles se puede observar el mismo patrón, por ejemplo, en *Nothofagus pumilio* el reclutamiento de plántulas fue inferior al 1.5 % [@torres2015seed]. En general los patrones de germinación *in situ* varían por grupos taxonómicos, distribución y condiciones ecológicas [@iralu2019ecology].
Como es bien conocido en el campo, la germinación de semillas en las orquídeas no es sencilla ya que depende de la disponibilidad de micorrizas [@rasmussen1995terrestrial]. Si bien existe controversia, hay especificidad de las orquídeas por ciertas especies de hongos micorrízicos, ya que hay evidencias de que ciertas especies de orquídeas presentan dicha especificidad [@balducci2024narrow], en tanto que otras no la presentan [@petrolli2022spatial], es indiscutible que para la germinación de las semillas de las orquídeas, las cuales carecen de endospermo, la asociación con un hongo micorrízico es indispensable para su germinación [@rasmussen2015germination]. Pero la germinación no sólo depende de esta asociación ya que muchas variables pueden influir incluyendo las condiciones bióticas y abióticas [@callaway2002epiphyte; @rasmussen2015germination; @gonzalez2024host].
En el siguiente gráfico del ciclo de vida emergido, se puede observar que ninguna de las plántulas sobrevive o crece a la siguiente etapa. Tenga en cuenta que en la primera columna de la matriz de transición todos los valores son cero. La población puede haber comenzado con muchas (incluso miles o cientos de miles) de plántulas, pero ninguna creció hasta convertirse en un juvenil o permaneció como plántula antes del siguiente muestreo. Esto da como resultado una matriz que es reducible *isIrreducible* = *FALSE* y por consecuencia no cumple con los requisitos necesarios para muchos de los análisis de matrices poblacionales.
```{r}
#| docx_fit: true
#| label: fig-sin_sentido_8
#| fig-cap: "Ciclo de vida correspondiente a la matriz Sp1matU_NS con estadios plántula, juvenil y adulto, sin transición desde plántulas (primera columna nula)."
Sp1matU_NS <- rbind(
c(0.0, 0.0, 0.0),
c(0.0, 0.7, 0.0),
c(0.0, 0.25, 0.995)
) # transition matrix
Sp1matF <- rbind(
c(0.0, 0.0, 1.0),
c(0.0, 0.0, 0.0),
c(0.0, 0.0, 0.0)
) # fertility matrix
Sp1matA_NS <- Sp1matU_NS + Sp1matF # TF matrix
stages <- c("plantula", "juvenil", "adulto")
plc_save(Sp1matU_NS, stages = stages, fontsize = 0, png = "images/sin_sentido_8_Sp1matU_NS.png")
isIrreducible(Sp1matU_NS)
```
::: callout-warning
## Cómo detectar y resolver — sobrevivencia cero en una etapa temprana
**Detección en R:**
``` r
which(colSums(matU) == 0) # ¿columnas enteras de ceros?
```
**Causa típica:** la primera etapa (semillas, plántulas) tiene una probabilidad de germinación o supervivencia genuinamente baja, y *ningún* individuo de los muestreados sobrevivió al siguiente censo. El valor observado es 0, pero el valor real no lo es.
**Cómo evitarlo o corregirlo:**
1. Aumentar el tamaño de muestra de la etapa crítica si es posible.
2. Acoplar muestreos *ex situ* (cajas de germinación, paquetes de malla) para estimar el reclutamiento con precisión separada del tamaño poblacional.
3. Usar un previo bayesiano biológicamente informado por la literatura (ver capítulo bayesiano y `raretrans`).
4. Si se decide reportar 0, declararlo como un límite inferior y reportar el tamaño de muestra que produjo esa estimación.
:::
------------------------------------------------------------------------
## Redondeo excesivo de valores
Uno de los errores más comunes en la construcción de los elementos de la matriz es el error de redondeo excesivo u otras estimaciones que dan como resultado valores de supervivencia superiores a 100%. Una estimación de supervivencia mayor a 1.00, genera que de manera artificial (por un milagro matemático) aumente la cantidad de individuos, ya que su origen no es debido a la reproducción (sexual u asexual), si no a un artificio matemático. El estimado de supervivencia de una etapa siempre tiene que ser menor o igual a 1. Tomemos el ejemplo del análisis de *Serapias cordigera* [@pellegrino2014effects] en donde los autores evaluaron las transiciones entre latencia, plántulas, roseta vegetativa y floración, en este ejemplo podemos observar que al sumar los valores de transición de la columna de plántulas de la matriz SerapiaU, obtenemos un valor de 1.01, lo que significa que no solamente las plántulas presentan una sobrevivencia perfecta, que ya vimos que es biológicamente imposible, si no que además están apareciendo plántulas de la nada (ya que las plántulas son incapaces de reproducirse sexual o asexualmente y por tanto su entrada en la matriz de fecundidad `SerapiaF` es cero), que provocan que la población de plántulas aumente en un 1% en cada período. Estos valores de sobrevivencia superiores a 1 son el resultado de errores en la recolección de datos y/o en el cálculo de la supervivencia de la etapa, pero más frecuentemente por el redondeo excesivo de los valores de sobrevivencia calculada al momento de incorporarlos a la matriz, en donde generalmente sólo se utilizan entre dos y tres decimales.
```{r}
#| docx_fit: true
#| label: fig-sin_sentido_9
#| fig-cap: "Ciclo de vida correspondiente a la matriz SerapiaA con estadios latente, plántulas, vegetativa y adulto, cuya primera columna suma más de 1."
SerapiaU <- rbind(
c(0.668, 0.122, 0.294, 0.401),
c(0.128, 0.000, 0.000, 0.000),
c(0.000, 0.302, 0.453, 0.366),
c(0.214, 0.364, 0.185, 0.207)
) # transition matrix
colSums(SerapiaU) # la suma de las columnas, nota que la primera columna es mayor de 100%.
SerapiaF <- rbind(
c(0.0, 0.0, 0.0, 0.0),
c(0.0, 0.0, 0.0, 0.0),
c(0.0, 0.0, 0.0, 0.0),
c(0.0, 0.0, 0.0, 0.0)
) # fertility matrix
SerapiaA <- SerapiaU + SerapiaF # TF matrix
stages <- c("latente", "plantulas", "vegetativa", "adulto")
plc_save(SerapiaA, stages = stages, png = "images/sin_sentido_9_SerapiaA.png")
# Tenga en cuenta que la suma de supervivencia para la etapa inicial es mayor de 1, igual a 1.01
```
::: callout-warning
## Cómo detectar y resolver — columna de U que suma más de 1
**Detección en R:**
``` r
which(colSums(matU) > 1) # ¿qué etapas tienen sobrevivencia > 100%?
round(colSums(matU), 4) # imprime las sumas con 4 decimales
```
**Causa típica:** redondeo a 2 ó 3 decimales en cada componente de la transición. Si las cuatro entradas que salen de una etapa se redondean independientemente, los errores se acumulan en el último decimal.
**¿Por qué importa?** Una columna que suma 1.01 en lugar de 1.00 implica un crecimiento artificial del 1 % por periodo —en 10 periodos, una sobreestimación de \$\approx\$10 % de $\lambda$ que no proviene de ninguna tasa vital biológica.
**Cómo evitarlo o corregirlo:**
1. Trabajar con al menos 4–5 decimales en los cálculos intermedios y reportar al menos 3 decimales en la matriz final.
2. Forzar la consistencia: calcular `mortalidad <- 1 - colSums(matU)` y verificar que es no-negativa.
3. Si la suma sigue excediendo 1, regresar al paso de cálculo de las tasas vitales —no es un problema de redondeo sino de doble conteo (un mismo evento entró en dos celdas).
:::
------------------------------------------------------------------------
## Estimaciones de fertilidad y ciclo de vida
La fecundidad influye considerablemente sobre los resultados de los modelos. Es por ello que cometer errores en la incorporación de valores, tanto en el ciclo de vida como en las matrices, puede dar lugar a interpretaciones sin sentido biológico. Por esa razón, en esta sección mostramos algunos ejemplos de errores al incorporar la fecundidad en el ciclo de vida y la matriz, y los problemas que generan. Vea el [capítulo de Fecundidad](#MatricesF) para más detalles.
### Ciclo de vida y etapa de fertilidad incorrectos
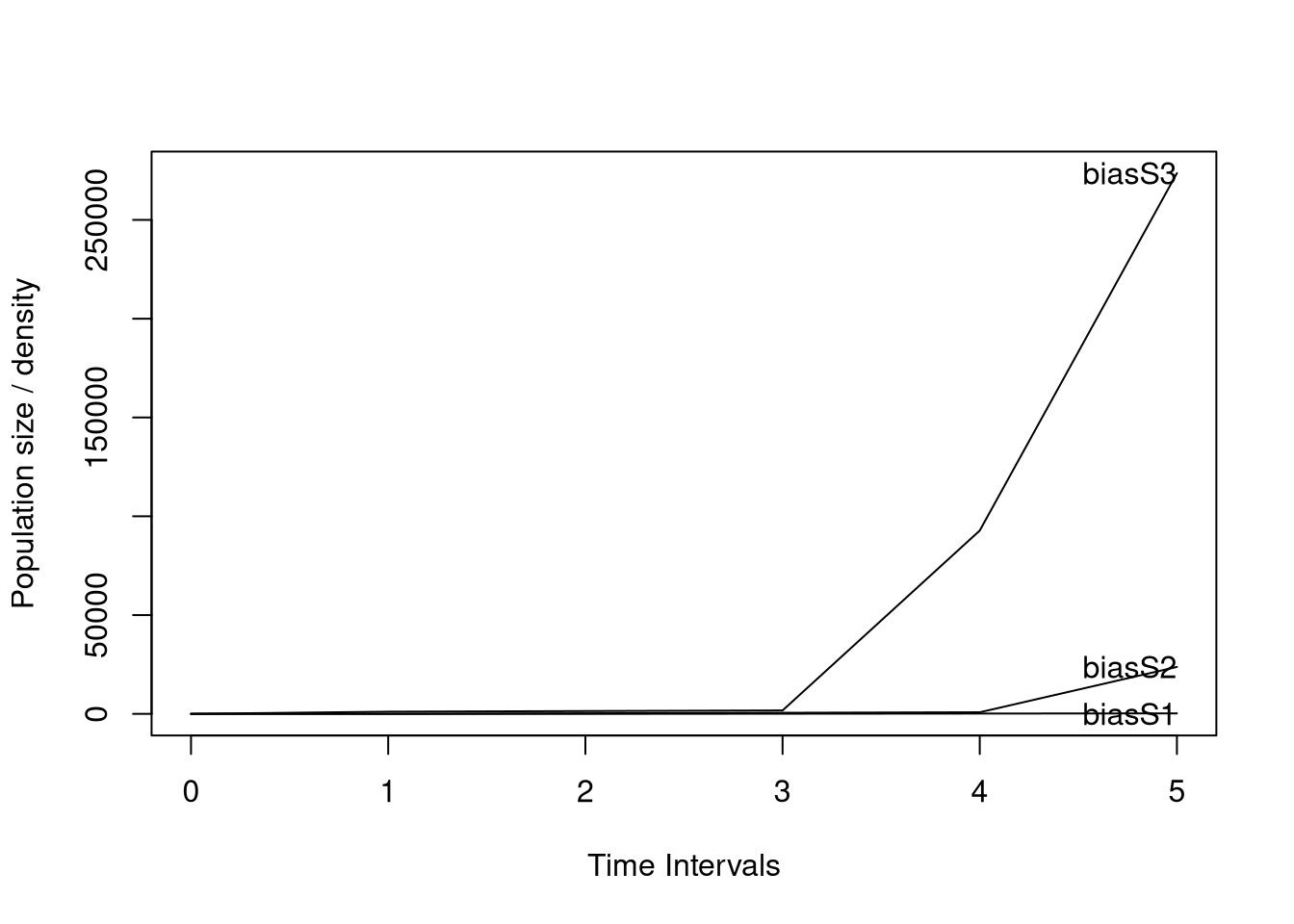
Suponga que tiene una especie en la que modela el ciclo de vida con los siguientes estadios: plántula, juvenil y adulto, en donde la fecundidad se calcula con base en el número de semillas producidas por un adulto (número medio de semillas por adulto). Después de analizar los datos y calcular los valores de transición (Sp1matU), se determinó que el número medio de semillas producido por adulto es 1100, a partir del cual se construyó la matriz de fecundidad (Sp1matF). Si agregamos a la matriz de transición la matriz de fertilidad, y a continuación evaluamos el valor de lambda y graficamos el crecimiento de la población con la función `project()`, tenemos un valor de lambda de 4.93 y después de tan solo 5 períodos de tiempo, el número de adultos supera los 250,000 individuos!!! Por consecuencia, la estimación de la tasa de crecimiento de la población sería incorrecta y engañosa, donde 30% de 11,000 semillas llegan a ser plántulas, un total de 3,300 plántulas!!!
Este error es el resultado del cálculo de la fecundidad, que debiera ser estimada con base en la primera etapa de la matriz, en este caso la etapa de plántula. Por lo tanto, la esquina superior derecha de la matriz de fertilidad no debe ser la **cantidad media de semillas** por planta, sino la **cantidad media de semillas que transitan a plántulas** producidas por una planta adulta. Recuerde que el número de semillas que llegan a plántula, son el resultado de múltiples factores; por ejemplo el porcentaje de viabilidad de las semillas por adulto, el porcentaje de semillas que arriban a un micro nicho propicio, el porcentaje de semillas que germinan y el porcentaje de sobrevivencia de las plántulas hasta el momento del muestreo, en donde se contabiliza el número de nuevas plántulas reclutadas. Por consecuencia el valor de esfuerzo reproductivo que entra en la matriz es condicional a todas estas condiciones.
```{r}
#| label: fig-sin_sentido_10
#| fig-cap: "Gráfico de la proyección poblacional (project con vector n y time 5) de la matriz Sp1matA_Fert con fecundidad de 1100 en el adulto."
Sp1matU_Fert <- rbind(
c(0.0, 0.0, 0.0),
c(0.3, 0.7, 0.0),
c(0.0, 0.25, 0.995)
) # matriz de transición
Sp1matF <- rbind(
c(0.0, 0.0, 1100.0),
c(0.0, 0.0, 0.0),
c(0.0, 0.0, 0.0)
) # matriz de fertilidad
Sp1matA_Fert <- Sp1matU_Fert + Sp1matF # TF matriz combinada: matA = matU + matF
stages <- c("plantula", "juvenil", "adulto")
lambda(Sp1matA_Fert)
# El cambio de tamaño de la población en 5 periodos de tiempo
n
pr <- project(Sp1matA_Fert, vector = "n", time = 5)
plot(pr) # Nota que después de solamente 5 años ya la población aumentó a 250,000 individuos
```
::: callout-warning
## Cómo detectar y resolver — fecundidad sobreestimada en la celda equivocada
**Detección:** en la matriz de fecundidad, la celda superior derecha contiene un número muy grande (cientos o miles) y produce $\lambda$ irreal (p. ej. \>2 en una especie longeva). Compare el reclutamiento real anual a la primera etapa con el valor en la matriz.
**Causa típica:** se anotó el número medio de **semillas producidas por adulto**, sin multiplicar por las probabilidades concatenadas de viabilidad, dispersión a un micrositio favorable, germinación y sobrevivencia hasta el primer censo.
**Cómo evitarlo o corregirlo:**
1. La entrada $f_{1,k}$ debe ser el número de **plántulas observadas reclutándose por adulto** (la primera etapa del modelo), no el número de semillas producidas.
2. Documente explícitamente cómo se calculó la fecundidad —ya sea contando reclutas directamente o multiplicando $\text{semillas} \times P_{\text{viabilidad}} \times P_{\text{germinación}} \times P_{\text{sobrevivencia plántula}}$ (ver el [capítulo de Fecundidad](#MatricesF)).
3. Especifique el tipo de censo (pre- o post-reproductivo) y si el modelo es birth-flow o birth-pulse (ver el [protocolo estandarizado del capítulo 121](#protocolo)).
4. Si quiere modelar la cadena completa, agregue una etapa de semillas explícita —pero entonces debe reportar la transición semilla → plántula, lo que en orquídeas es un dato muy difícil de obtener.
:::
------------------------------------------------------------------------
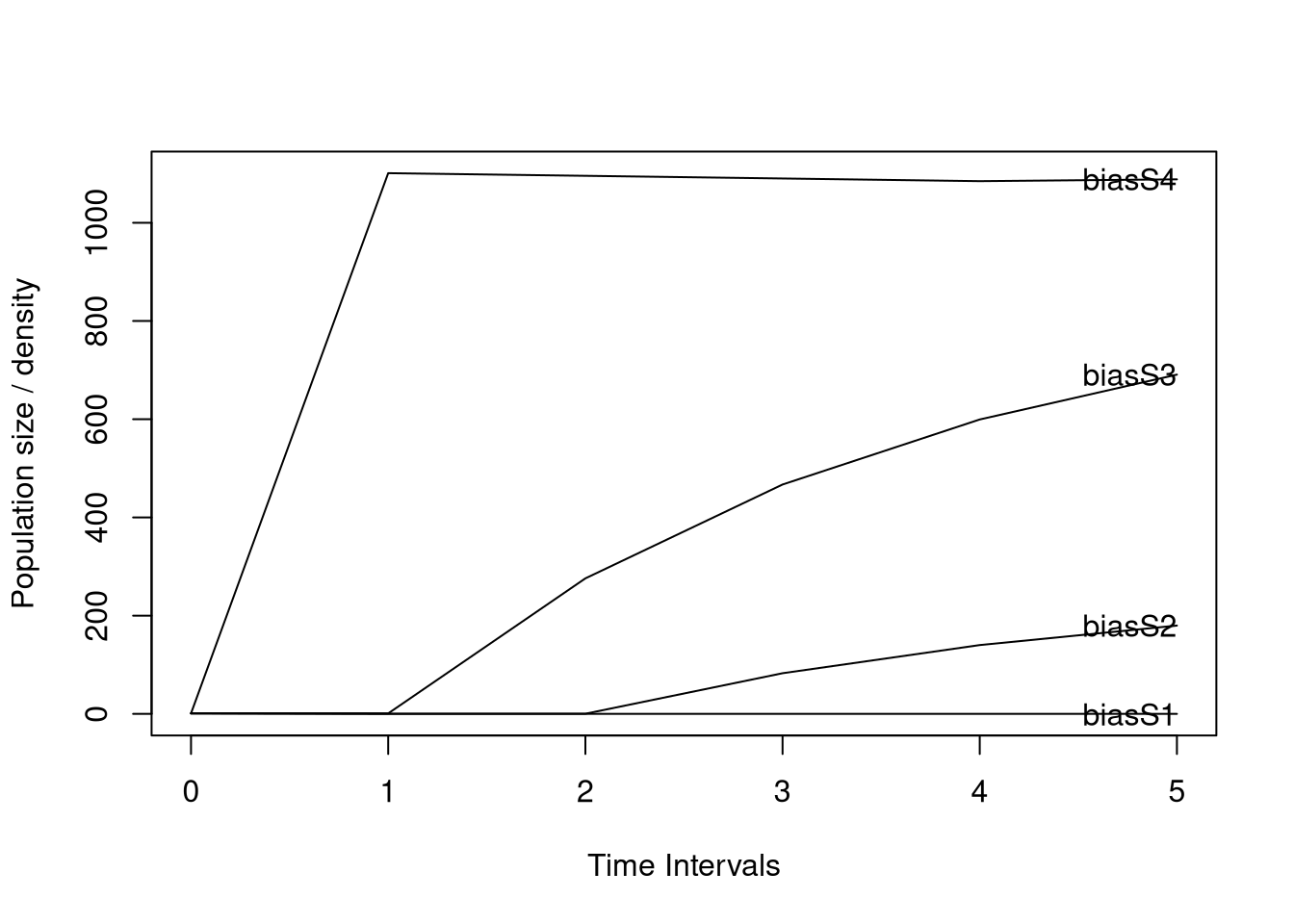
### Añadir una etapa de semillas
Si deseamos agregar una etapa de semilla a nuestro modelo, debemos incluir la etapa en nuestro modelo y crear una matriz con una nueva etapa *semilla*, para lo cual será necesario tener una estimación del número de semillas que germinan y se convierten en plántulas para poder calcular la transición de semillas a plántulas de nuestro modelo. En el caso de las orquídeas, dicha estimación es bastante difícil de lograr ya que las semillas de las orquídeas, debido a su tamaño minúsculo, son difíciles de seguir en la naturaleza. Aunque existen métodos para seguir las semillas como el de paquetes de malla de fitoplancton donde uno añade las semillas para monitorear esa etapa [@rasmussen1993seed; @batty2006situ; @shao2024improved] o algún método de huella genética para determinar la procedencia de las plántulas, que nos pueden ayudar a tener una estimación de esta transición, esta será incorrecta la mayoría de las veces, ya que en el caso de los paquetes de semillas se estará sobreestimando la transición ya que no se estaría considerando la pérdida de semillas por dispersión, en tanto que en el método de huella genética es muy probable que se subestime la transición ya que es muy difícil encontrar las plántulas en campo, sobre todo en orquídea epifitas. Por lo tanto, la estimación de la fertilidad en las orquídeas es un desafío y requiere un muestreo y análisis cuidadoso.
```{r}
#| docx_fit: true
#| label: fig-sin_sentido_11
#| fig-cap: "Ciclo de vida correspondiente a la matriz Sp1matA_Fert2 con estadios semillas, plántula, juvenil y adulto, con fecundidad de 1100 y transición semilla-plántula de 0.0001."
Sp1matU_Fert2 <- rbind(
c(0.0, 0.0, 0.0, 0.0),
c(0.0001, 0.0, 0.0, 0.0),
c(0.0, 0.3, 0.7, 0.0),
c(0.0, 0.0, 0.25, 0.995)
) # transition matrix
Sp1matF2 <- rbind(
c(0.0, 0.0, 0.0, 1100.0),
c(0.0, 0.0, 0.0, 0.0),
c(0.0, 0.0, 0.0, 0.0),
c(0.0, 0.0, 0.0, 0.0)
) # fertility matrix
Sp1matA_Fert2 <- Sp1matU_Fert2 + Sp1matF2 # TF matrix
stages <- c("semillas", "plantula", "juvenil", "adulto")
plc_save(Sp1matA_Fert2, stages = stages, png = "images/sin_sentido_11_Sp1matA_Fert2.png")
lambda(Sp1matA_Fert2)
# mostrar el cambio en el tamaño poblacional
pr <- project(Sp1matA_Fert2, vector = "n", time = 5)
plot(pr) # Nota que ahora el número de adultos no aumentó al nivel del modelo anterior
```
------------------------------------------------------------------------
### Semillas latentes
En el ejemplo anterior no hemos incluido una etapa de latencia de semillas. En otras palabras: ¿las semillas de orquídeas pueden sobrevivir por un periodo de tiempo largo? Esto sería tener un banco de semillas. ¿O las semillas, si no germinan el primer año, mueren? En muchas especies de plantas, las semillas pueden permanecer latentes durante uno o más años. No se sabe si las semillas de las orquídeas están inactivas durante mucho tiempo [@gale2010constraints; @gale2021international], excepto para algunas especies que han demostrado que las semillas aún están vivas después de varios años en el suelo usando la prueba de tinción de tetrazolio [@rasmussen1993seed; @whigham2006seed]. Es posible que las orquídeas terrestres tengan un banco de semillas, pero no se ha demostrado que las orquídeas epífitas tengan un banco de semillas. Por lo tanto, si se desea incluir una etapa de latencia de semillas en el modelo, es necesario tener una estimación del número de semillas que permanecen latentes y que pueden germinar en el futuro.
------------------------------------------------------------------------
## Problemas de análisis de datos
### Estimaciones de intervalos de confianza incorrectas
Las estimaciones de dispersión en los parámetros de la matriz (supervivencia, transición, muerte y fertilidad) son útiles por múltiples razones tal como reconocer la confianza que se debería tener sobre el estimado puntual (el promedio o mediana). Por ello los parámetros con gran dispersión deben verse con precaución. El enfoque más básico es comprender la dispersión en el valor del parámetro como una función del tamaño de la muestra y el efecto espacial o temporal. Los estimados de dispersión pueden ser útiles para simulaciones y para comprender la incertidumbre en los estimados puntales y el comportamiento de la población como son lambda y la probabilidad de persistencia y extinción.
Los parámetros de supervivencia, muerte, permanencia y transición no se distribuyen normalmente ya que sus valores van de cero a uno. Ningún valor puede ser menor de cero o mayor de uno, incluidos los intervalos de confianza (limitados por 0 y 1). Si se usa la distribución gaussiana (distribución normal) es probable que el intervalo de confianza esté fuera de los límites.
Supongamos que deseamos calcular la probabilidad de supervivencia de una etapa y sus intervalos de confianza del 95%. Primeramente se determinó que de los 20 individuos muestreados uno falleció y 19 sobrevivieron entre el primer muestreo y el segundo muestreo. Posteriormente, se estimó un intervalo de confianza del 95% de la proporción de individuos que murieron basándose en una distribución gaussiana.
Donde la proporción que murió es $\hat{p}$ y el número que murió es $n_d$ y $n$ es el tamaño de la muestra. En nuestro caso $n_d=1$ y $n=20$.
- Construya un intervalo de confianza del 95% de la proporción de individuos que murieron basándose en una distribución gaussiana.
Donde la proporción que murió es $\hat{p}$ y el número que murió es $n_d$ y $n$ es el tamaño de la muestra
$$\hat{p}=\frac{n_d}{n}$$ con una probabilidad de muerte del 5%.
```{r sin_sentido_12}
p <- 1 / 20
p
```
El IC del 95% de una proporción se calcula usando la siguiente fórmula si se asume una distribución normal
$$\hat{p} \pm Z_{0.05} \sqrt{\frac{\hat{p}(1-\hat{p})}{n}}$$
- $Z_{0.05}$ es el valor crítico de $Z$ para un IC del 95% = 1.96. Nota que, con ese cálculo, uno de los intervalos de confianza resulta negativo, lo cual es ilógico (una proporción no puede ser negativa).
```{r sin_sentido_13}
n <- 20
HCI <- p + (1.96 * sqrt((p * (1 - p)) / n)) # IC alto
HCI
LCI <- p - (1.96 * sqrt((p * (1 - p)) / n)) # IC bajo
LCI # Nota que el valor es sin sentido, ya que es negativo (¿qué es una proporción negativa?)
```
Un método más fácil es usar la función R `propCI()` de la `library(interpretCI)` (Script 12). La función `propCI()` calcula el intervalo de confianza del 95% de una proporción binomial. La función requiere el tamaño de la muestra, el número de eventos y el nivel de significancia. En este caso, el tamaño de la muestra es 20, el número de eventos es 1 (un individuo fallecido) y el nivel de significancia es 0.05. Note que el intervalo de confianza incluye valores negativos (**irreales**). Para resolver esto hay que usar funciones estadísticas alternativas como la distribución de Dirichlet en donde no se asume una distribución normal.
```{r sin_sentido_14}
resultado <- propCI(n = 20, p = 0.05, alpha = 0.05)
resultado
```
::: callout-warning
## Cómo detectar y resolver — IC fuera de \[0, 1\]
**Detección en R:** los límites del IC reportan valores `< 0` o `> 1`, o un error estándar mayor que la propia probabilidad cuando $\hat p$ está cerca de 0 o de 1.
**Causa típica:** la fórmula gaussiana $\hat p \pm z \sqrt{\hat p (1-\hat p)/n}$ supone que la distribución muestral de $\hat p$ es simétrica, lo cual no es cierto cerca de las fronteras. Con $\hat p = 0.05$ y $n = 20$, el error estándar gaussiano es similar a $\hat p$, y el límite inferior se vuelve negativo.
**Cómo evitarlo o corregirlo:** usar métodos que respeten los límites \[0, 1\]:
1. **Wilson o Clopper–Pearson** (frecuentista): `binom::binom.confint(x = 1, n = 20, methods = c("wilson", "exact"))`.
2. **Beta–Binomial** para una sola probabilidad (bayesiano): la posterior conjugada con un previo Beta(1,1) es Beta($x+1$, $n-x+1$).
3. **Dirichlet–multinomial** cuando hay más de dos categorías —es la opción correcta para una columna completa de la matriz U, donde estasis, transición y mortalidad deben sumar 1 simultáneamente. Esto es lo que se desarrolla en la siguiente sección.
:::
------------------------------------------------------------------------
## Distribución Dirichlet de intervalo de confianza
Sin embargo, ¿cómo se calcula el IC del 95% cuando hay más de dos proporciones? Debido a que la proporción de todas las etapas depende de la proporción de las otras etapas, el análisis debe considerar la proporción y el IC del 95% debe incluir todas las etapas simultáneamente. La función requerida para esto es la función de Dirichlet multinomial (multigrupo).
La distribución de Dirichlet es una distribución de probabilidad continua multivariada que se utiliza para modelar la distribución de proporciones en un espacio de probabilidad. La distribución de Dirichlet es una generalización de la distribución beta a más de dos categorías. La distribución beta está limitada a valores de cero a uno inclusivo. La distribución de Dirichlet se utiliza comúnmente en el análisis de datos de proporciones, donde las proporciones de las categorías deben sumar uno.
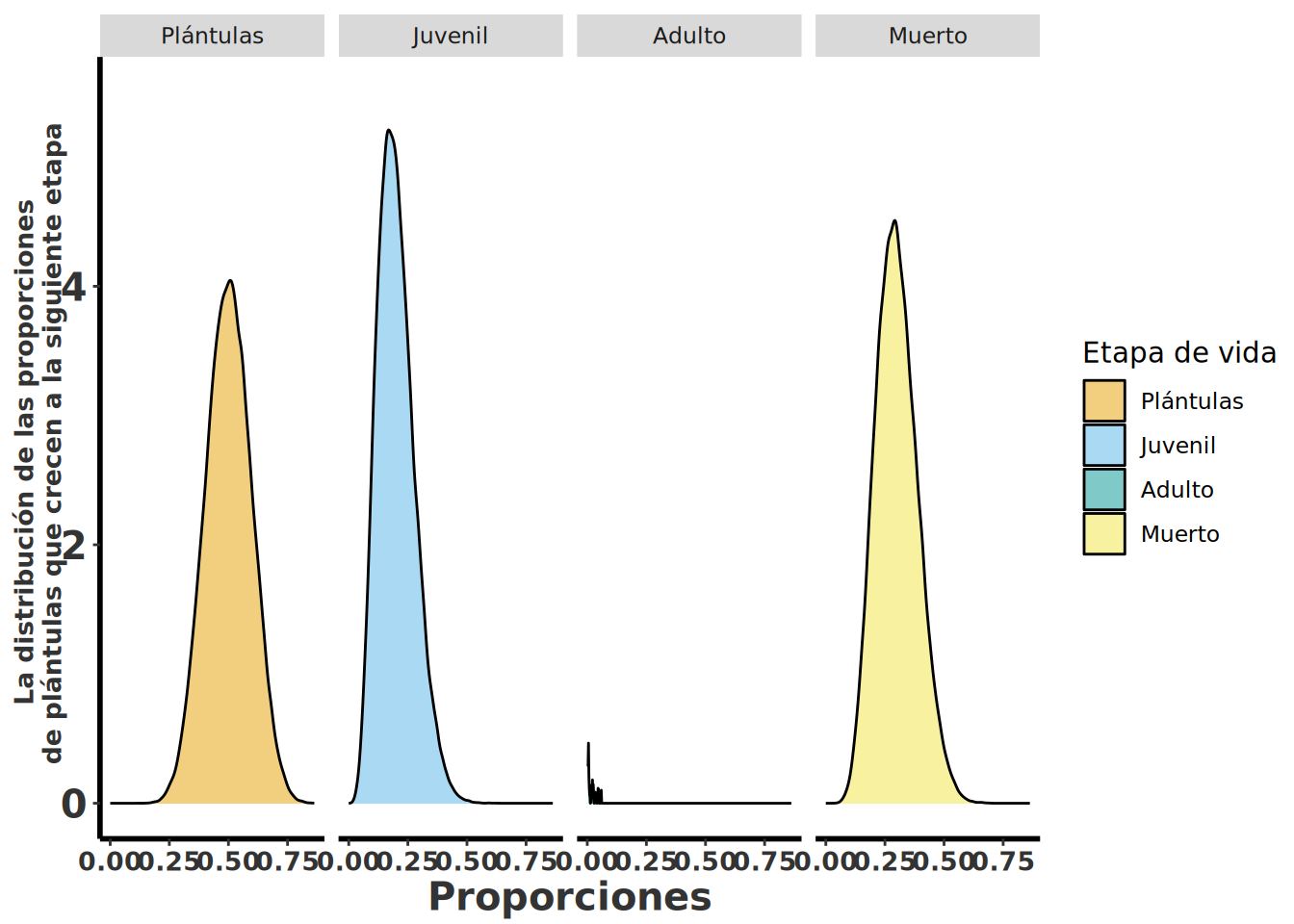
En el siguiente script estimamos los intervalos de credibilidad del 95% de la transición y estasis de plántulas a plántulas (0.5), juveniles (0.2), adultos (0) y muerte (0.30). Tenga en cuenta que en el gráfico del ciclo de vida, como de costumbre, se excluyen las probabilidades de muerte. Esta función es un acercamiento bayesiano por consecuencia es un intervalo de credibilidad (ICr) y no un intervalo de confianza (IC). La diferencia entre ambos es que el intervalo de credibilidad es una estimación bayesiana, mientras que el intervalo de confianza es una estimación frecuentista.
```{r, message=FALSE}
#| docx_fit: true
#| label: fig-sin_sentido_15
#| fig-cap: "Ciclo de vida correspondiente a la matriz Dirichlet con estadios plántula, juvenil y adulto, sin retención en plántula ni juvenil."
Dirichlet <- rbind(
c(0.5, 0.0, 0.0),
c(0.2, 0.0, 0.0),
c(0.0, 0.3, 0.7)
) # transition matrix
stages <- c("plantula", "juvenil", "adulto")
plc_save(Dirichlet, stages = stages, png = "images/sin_sentido_15_Dirichlet.png")
```
Para evaluar el intervalo ICr del 95% simultáneamente de las cuatro transiciones (plántulas a plántulas, plántulas a juvenil, plántulas a adultos y plántulas a muerte), usamos la `library(MCMCpack)`, que es un paquete dedicado a realizar simulaciones de Monte Carlo de Cadenas Markovianas (*Markov Chain Monte Carlo*). Para lo cual usaremos la función `MCmultinomdirichlet`. Tenga en cuenta que la primera lista son las proporciones que se muestran en la matriz para la etapa de plántula `c(.50*n, .2*n, .0*n, .3*n)` multiplicadas por el tamaño de la muestra `n = 25`. La segunda lista son las previas bayesianas `c(0.5, 0.2, 0.0001, 0.2999)`; en este caso asumimos que la suma de las previas es igual a 1. Esto da como resultado una confianza muy baja en la percepción previa de lo que son las transiciones — estamos dando muy poco peso a la percepción previa en los análisis. Otra opción de previa para la transición es que sean iguales; podríamos haber usado `c(1, 1, .0001, 1)`, donde esta previa sugiere una transición igual para todos los estados menos para la transición de plántula a adulto. IMPORTANTE: las previas deberían ser **biológicamente realistas**, usando lo conocido de la biología de los organismos que se estudian. Por experiencia y usando la literatura se puede hacer un estimado de las transiciones y usarlo como previa. El concepto de lo anterior no se puede explicar completamente aquí; consulte las siguientes referencias [@tremblay2021population; @van2021bayesian].
Los resultados en la figura muestran que hay mucha dispersión alrededor de la proporción media de las estasis y las transiciones, como se esperaba debido al pequeño tamaño de la muestra.
- Hacemos una simulación de 100,000.
- Recolectamos los resultados en la variable **L**.
- Convertimos esta lista en un data frame.
- Reorganizamos el data frame para que sea más fácil de graficar con la función `stack()`.
- Añadimos una columna con el tamaño de muestra, n=25.
- Cambiamos los nombres de las etapas de la vida para que sean más fáciles de leer en la gráfica.
- Graficamos los resultados
```{r sin_sentido_16, message=FALSE}
n <- 25
L <- posteriorPRIORL <- MCmultinomdirichlet(
c(.50 * n, .2 * n, .0 * n, .3 * n),
c(.5, .2, 0.0001, .3),
mc = 100000
)
dfL <- as.data.frame(L)
t(summary(dfL))
# head(dfL)
stack_dfL <- stack(dfL)
comb_dfbL <- cbind(stack_dfL, T = "25")
All_Data3 <- comb_dfbL
levels(All_Data3$ind)[levels(All_Data3$ind) == "pi.1"] <- "Plántulas"
levels(All_Data3$ind)[levels(All_Data3$ind) == "pi.2"] <- "Juvenil"
levels(All_Data3$ind)[levels(All_Data3$ind) == "pi.3"] <- "Adulto"
levels(All_Data3$ind)[levels(All_Data3$ind) == "pi.4"] <- "Muerto"
```
- Graficamos los resultados
```{r}
#| label: fig-sin_sentido_17
#| fig-cap: "Gráficas de densidad (geom_density) con facetas por estadio que muestran la distribución de las proporciones de plántulas que crecen a la siguiente etapa."
ggplot(data = All_Data3, aes(x = values, fill = ind)) +
geom_density(alpha = .5) +
scale_y_continuous(limit = c(0, 5.5)) +
scale_colour_hue(l = 60) +
facet_grid(~ind) +
ylab(
"La distribución de las proporciones \n de plántulas que crecen a la siguiente etapa"
) +
xlab("Proporciones") +
labs(fill = "Etapa de vida") +
rlt_style_fill() +
theme(axis.text.x = element_text(angle = 45, hjust = 1))
```
Ahora calculemos el ICr del 95% de las transiciones
Ahora que los datos están organizados en un *data frame* es fácil extraer los valores de las transiciones y calcular los intervalos de credibilidad del 95% de las transiciones, estasis y supervivencia. Para ello usamos la función `ddply` del paquete `plyr` para resumir los datos por la columna `ind` (etapa de vida) y calcular el promedio, desviación estándar, mediana, cuantil 5% y cuantil 95% de los valores de cada etapa.
- Intervalos de credibilidad de transiciones, estasis y supervivencia basados en simulación y distribución de Dirichlet.
Aspectos importantes a tener en cuenta.
- los ICr del 95% están acotados entre 0 y 1. Los valores por debajo de cero o por encima de uno en los parámetros no tendrían sentido.
- la suma de los promedios es igual a 1.
- la forma de la distribución no se distribuye normalmente, ver figura anterior. La forma de la distribución se llama distribución beta.
```{r sin_sentido_18}
Transiciones <- ddply(
All_Data3,
c("ind"),
summarise,
mean = round(mean(values), 4),
sd = round(sd(values), 5),
median = round(median(values), 4),
# sem = round(sd(values)/sqrt(length(values)),6), # para calcular el error estándar de la media
ICr5 = round(quantile(values, probs = c(0.05)), 4), # el cuantil 5
ICr95 = round(quantile(values, probs = c(0.95)), 4), # el cuantil 95
min = min(values),
max = max(values)
)
Transiciones
```
Para visualizar los intervalos de credibilidad de las otras transiciones basta con cambiar las variables de la función `L` y `n`. En este caso, cambiamos el tamaño de la muestra correspondiente a esta etapa.
------------------------------------------------------------------------
### Tamaño de muestra y estimaciones de intervalos de confianza con n2=250
Para comparar con el ejemplo anterior asumimos que en lugar de tener solamente 25 plántulas teníamos 250 para las estimaciones.
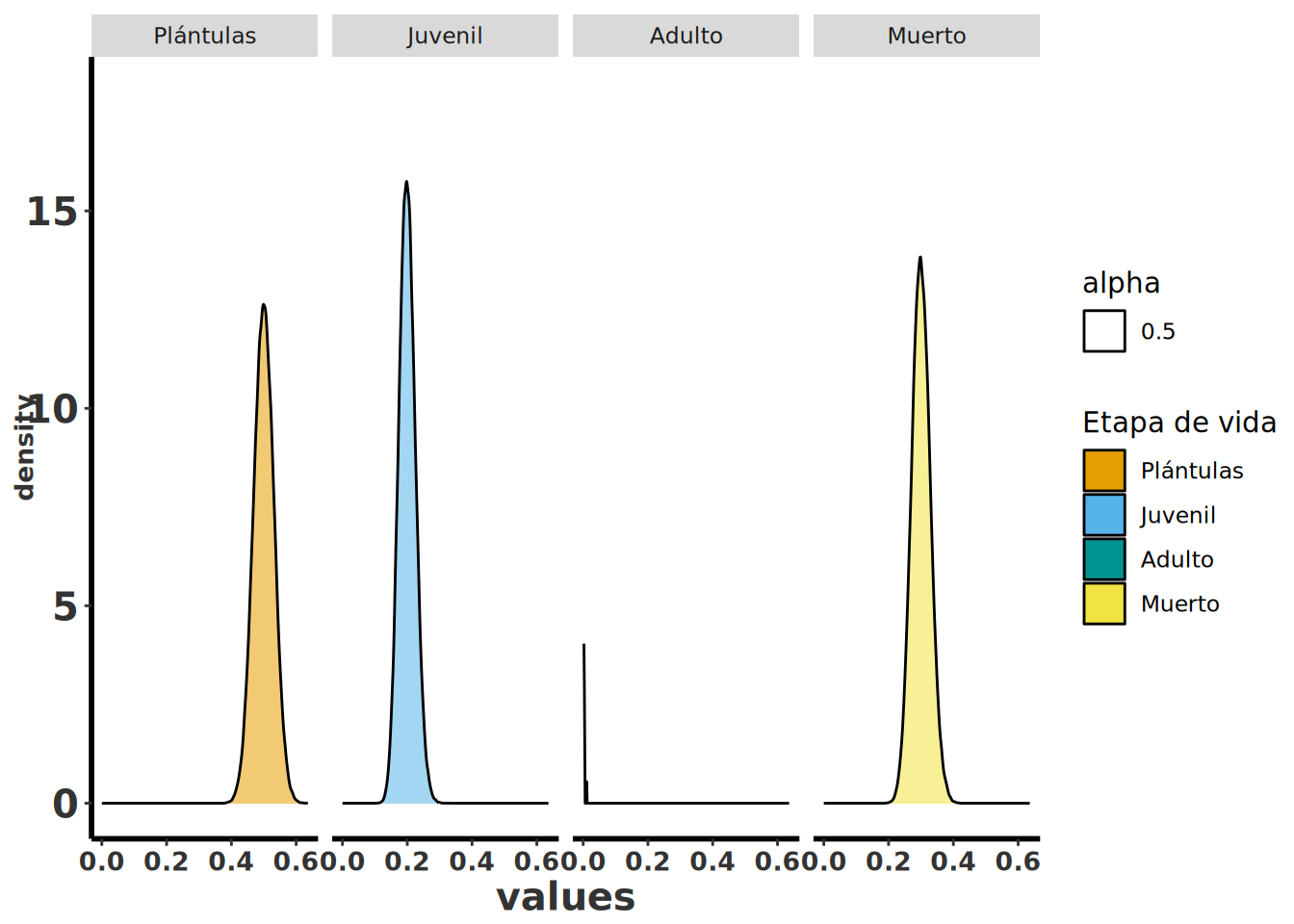
Note que con los mismos estimados puntuales pero con un tamaño de muestra mayor la mediana sigue en el mismo lugar (casi) pero los ICr cambian mucho y son más reducidos los anchos de la dispersión, ya que con más datos en el muestreo existe más confianza resultado de una reducción en el ICr. Este análisis es primordial para entender la confianza que uno debería tener sobre los estimados de los parámetros de la matriz. Si se observa un ICr muy grande, se recomendaría modificar la recolección de datos en el campo para aumentar el tamaño de muestra de esa etapa específica.
```{r}
#| label: fig-sin_sentido_19
#| fig-cap: "Gráficas de densidad (geom_density) con facetas por estadio (Plántulas, Juvenil, Adulto, Muerto) de las proporciones simuladas con MCmultinomdirichlet en All_Data4."
n2 <- 250
L <- posteriorPRIORL <- MCmultinomdirichlet(
c(.50 * n2, .2 * n2, .0 * n2, .3 * n2),
c(.5, .2, 0.0001, .3),
mc = 100000
)
dfL <- as.data.frame(L)
t(summary(dfL))
# head(dfL)
stack_dfL <- stack(dfL)
comb_dfbL <- cbind(stack_dfL, T = "25")
All_Data4 <- comb_dfbL
levels(All_Data4$ind)[levels(All_Data4$ind) == "pi.1"] <- "Plántulas"
levels(All_Data4$ind)[levels(All_Data4$ind) == "pi.2"] <- "Juvenil"
levels(All_Data4$ind)[levels(All_Data4$ind) == "pi.3"] <- "Adulto"
levels(All_Data4$ind)[levels(All_Data4$ind) == "pi.4"] <- "Muerto"
ggplot(data = All_Data4, aes(x = values, fill = ind)) +
geom_density(aes(alpha = .5)) +
scale_y_continuous(limit = c(0, 18)) +
scale_colour_hue(l = 60) +
facet_grid(~ind) +
labs(fill = "Etapa de vida") +
rlt_style_fill() +
theme(axis.text.x = element_text(angle = 45, hjust = 1))
```
### Los estimados de intervalo de credibilidad con un tamaño de muestra de n=250
Note que ahora los intervalos de credibilidad son más estrechos y no son negativos ni mayores de uno.
```{r sin_sentido_20}
Transiciones <- ddply(
All_Data4,
c("ind"),
summarise,
mean = round(mean(values), 3),
sd = round(sd(values), 4),
median = median(values),
# sem = round(sd(values)/sqrt(length(values)),6),
CI5 = quantile(values, probs = c(0.05)),
CI95 = round(quantile(values, probs = c(0.95)), 4),
min = min(values),
max = max(values)
)
Transiciones
```