Código
library(tidyverse)
library(ggtext)
library(janitor)
library(popbio)
library(raretrans)
library(ggpubr)
library(flextable)Los métodos de simulación permiten explorar la dinámica poblacional bajo escenarios variables y condiciones no observadas directamente en los datos. Este capítulo introduce las simulaciones poblacionales como herramientas prospectivas para evaluar trayectorias futuras, variabilidad demográfica y respuestas a cambios en los procesos vitales.
Por: Raymond L. Tremblay
Las simulaciones poblacionales constituyen una extensión natural de los modelos matriciales al permitir proyectar poblaciones bajo condiciones variables, estocásticas o dependientes del tiempo. A diferencia de los análisis analíticos que se centran en el comportamiento asintótico, las simulaciones permiten explorar trayectorias transitorias, evaluar la variabilidad demográfica y analizar respuestas poblacionales ante cambios explícitos en los procesos vitales.
En este capítulo se presentan los fundamentos conceptuales de las simulaciones poblacionales, mostrando cómo pueden implementarse a partir de matrices de proyección y transiciones demográficas previamente definidas. Se discuten distintos tipos de simulaciones, incluyendo simulaciones determinísticas, estocásticas y basadas en escenarios, así como sus aplicaciones para evaluar extinción, recuperación y efectos de manejo.
Asimismo, se analiza el papel de las simulaciones como herramientas complementarias a enfoques analíticos como la elasticidad y los LTRE. Mientras estos últimos permiten identificar la importancia relativa o las causas de diferencias observadas, las simulaciones permiten evaluar qué podría ocurrir bajo condiciones alternativas. Este capítulo se diferencia de los anteriores porque se enfoca en la exploración explícita del futuro poblacional, integrando estructura demográfica, variabilidad y supuestos de manejo para generar predicciones y apoyar la toma de decisiones en ecología y biología de la conservación.
Las simulaciones se construyen a partir de las transiciones demográficas introducidas en el capítulo de Transiciones. Las trayectorias simuladas se interpretan en relación con la tasa de crecimiento poblacional discutida en el capítulo de Crecimiento poblacional. A diferencia de los análisis LTRE, que explican diferencias observadas, las simulaciones permiten explorar escenarios futuros no observados. Las simulaciones pueden extenderse naturalmente dentro de un marco bayesiano, como se describe en el capítulo de Acercamiento bayesiano.
library(tidyverse)
library(ggtext)
library(janitor)
library(popbio)
library(raretrans)
library(ggpubr)
library(flextable)rlt_theme <- theme(
text = element_text(family = "Libertinus Serif"),
axis.title.y = element_text(colour = "grey20", size = 10, face = "bold"),
axis.text.x = element_text(colour = "grey20", size = 10, face = "bold"),
axis.text.y = element_text(colour = "grey20", size = 15, face = "bold"),
axis.title.x = element_text(colour = "grey20", size = 15, face = "bold")
) +
theme(
# Remover los bordes
panel.border = element_blank(),
# Remover las líneas
panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
# Remover el fondo del panel
panel.background = element_blank(),
# añadir lineas más gruesas
axis.line.x = element_line(colour = "black", linewidth = 1),
axis.line.y = element_line(colour = "black", linewidth = 1)
)
diverging <- c("#E69F00", "#56B4E9", "#009392", "#F0E442", "#0072B2", "#D55E00", "#CC79A7", "#000000")
# --- 2. Combinarlos en un objeto de tipo lista ---
# Esta lista se puede añadir al gráfico con un solo '+'
rlt_style_fill <- function() {
list(
rlt_theme,
scale_fill_manual(values = diverging)
)
}
rlt_style_colour <- function() {
list(
rlt_theme,
scale_colour_manual(values = diverging)
)
}A continuación se describen los procedimientos para implementar simulaciones poblacionales a partir de matrices de proyección y transiciones demográficas, incluyendo ejemplos determinísticos y estocásticos.
El término biología pudiese ser un sinónimo de variación biológica. No hay dos organismos iguales, ni siquiera gemelos/clones que sean completamente idénticos. La variación biológica es una característica fundamental de la vida y es la base de la evolución; no puede haber evolución sin variación. Cuando consideramos los individuos, por ejemplo humanos, fácilmente reconocemos variación —en tamaño, color de ojos, susceptibilidad a cáncer o infección, y efecto del ambiente (por ejemplo la dieta o acceso a la educación)— entre muchos factores que impactan la supervivencia, crecimiento y reproducción. Pero desafortunadamente cuando consideramos los animales y plantas de una misma especie, frecuentemente se obvia la variación en los análisis. Podemos observar variación a múltiples niveles en una misma especie:
Todos estos componentes de variación tendrán impacto en la dispersión de los estimados de los parámetros de la matriz de transición y el crecimiento poblacional, además del comportamiento de la población en el tiempo. Hay un componente que frecuentemente se deja en el olvido: el efecto de la variación por tamaño de muestra pequeño sobre los estimados, que llamamos estocasticidad demográfica. En este capítulo se discute el efecto de la variación en tiempo, espacio y tamaño de muestra sobre los estimados de los parámetros de la matriz y el crecimiento poblacional.
La variación sub-individual es la variación que se observa en un individuo. Por ejemplo, la variación en el tamaño de hojas, flores, frutos, semillas dentro del mismo individuo o la cantidad de hojas, flores, frutos, semillas en tiempos distintos. Este tipo de variación es común en plantas y es un componente importante de la variación total pero raramente se considera en los análisis. Carlos Herrera ha demostrado claramente que este componente no solamente es parte de la variación total de las especies, sino que es importante para entender los procesos ecológicos y evolutivos (Herrera 2009). Además, cuando uno compara la variación total con la variación sub-individual, la variación sub-individual es mucho más grande que la variación entre individuos y poblaciones. Esta variación, al conocimiento de los presentes autores, nunca ha sido incluida en análisis de dinámica poblacional ni integrada dentro de un MPP. En un estudio del tamaño de las flores en Lepanthes rupestris se observó que la variación en el tamaño de flores disminuye a lo largo de la inflorescencia y que la probabilidad de remoción de pollinia y de deposición de estas sobre el estigma no siguen el mismo patrón: la probabilidad de remoción de pollinia aumenta con flores más pequeñas, pero la probabilidad de frutos disminuye (Tremblay 2006). Por consecuencia, cuando se recolecta la información sobre el esfuerzo reproductivo de una especie, puede ser que la variación sub-individual impacte los estimados de la matriz de transiciones, debido a que las flores en este periodo eran de diferentes tamaños. Las inflorescencias de Lepanthes pueden ser activas produciendo flores por meses y años.
La variación entre individuos es la diferencia que se observa entre individuos de una misma población. Por ejemplo, la variación en el tamaño de los individuos, cantidad de hojas, cantidad de flores, cantidad de frutos entre individuos de una misma población influye en la probabilidad de sobrevivir, crecer y reproducirse. La variación entre individuos de diferentes tamaños o etapas impacta grandemente la supervivencia, crecimiento y reproducción de los individuos. Los trabajos de Harper y otros han demostrado que esa variación es común en las plantas (Harper 1977) y típicamente es más importante que la edad de los individuos en la mayoría de las plantas. En orquídeas, un ejemplo de esa variación es que la mayoría de los individuos reproductivos en una población no produce frutos (Ackerman et al. 2020).
La variación entre poblaciones es la variación que se observa entre grupos de individuos de una misma especie en diferentes sitios. Poblaciones distintas pudiesen ser influenciadas por las diferencias abióticas e interacciones bióticas únicas a cada población. Esa variación pudiese influenciar la dinámica de cada población y la dinámica de la especie en general. Las diferencias entre poblaciones pudiesen ser influenciadas por condiciones específicas como la variación genética entre poblaciones, variación abiótica (recursos como el agua, nitrógeno, luz), variación en la interacción biótica (e.g. micorriza, diversidad y cantidad de polinizadores, tipo de forófitos), variación en la historia de la población (procesos de sucesión), entre otros factores, además de la plasticidad fenotípica que se define como la interacción del ambiente sobre la expresión genética (Nicotra et al. 2010). Sin duda, las variables abióticas y bióticas pueden influenciar las transiciones, estasis y reproducción de forma diferencial entre las diferentes etapas del ciclo de vida de la especie.
Este componente de variación incluye la variación típicamente relacionada a la variación abiótica que cambia de un periodo de tiempo a otro; por ejemplo, el efecto de la cantidad de lluvia, número de días sin lluvia, temperatura, entre muchas otras variables. Pero no se debería limitar a considerar solamente la variación abiótica en el tiempo, ya que la variación biótica también puede cambiar de un periodo de tiempo a otro. Por ejemplo, la densidad y/o composición de especies en una comunidad cambia de un periodo de tiempo a otro, e incluso la variación de patógenos y otras interacciones bióticas alrededor de la especie de interés. Todo lo anterior pudiese influenciar la dinámica y los estimados de los parámetros de la matriz. Por ejemplo, considera la variación en la presencia de herbívoros o patógenos que cambia de un periodo de tiempo a otro, resultando en variación en la competencia interespecífica. Por consecuencia, la variación en el tiempo es mucho más que la variación abiótica en el tiempo. Allí, uno también tiene que incluir el efecto de bonanzas (eventos raros positivos, por ejemplo: reclutamiento excepcional en algunos años) (Antonini et al. 2020) y catástrofes (eventos raros negativos, por ejemplo: huracanes, mortandad de un hospedero) (Hiernaux et al. 2009), que pudiesen influenciar la dinámica de la especie de interés. Un ejemplo drástico de su impacto es la mortandad casi completa de epífitas al pasar huracanes fuertes (Goode y Allen 2008), o que el efecto sea diferencial entre especies o sustratos (Rodrı́guez-Robles et al. 1990).
La gran mayoría de los estudios usando MPP son para evaluar la probabilidad de supervivencia en una especie en peligro de extinción. La razón principal de estos estudios es que quedan muy pocos individuos en la población o pocas poblaciones, y esas poblaciones se consideran en riesgo.
Por ejemplo si tuviésemos en una población solamente de un adulto y este adulto sobrevive de un año a otro entonces la probabilidad de supervivencia es 1.0. Pero si tuviésemos solamente un adulto y este adulto muere entonces la probabilidad de supervivencia es 0.0. Las probabilidad de supervivencia pueden ser solamente 1.0 o 0.0 respectivamente y otros valores no son posibles, pero la interpretación biológica es sesgada, porque ya hay solamente dos alternativas. Pero si tuviésemos 1,000 o 10,000 individuos es muy poco probable que todos se mueren o todos sobreviven, y hay muchas alternativas posibles. Este es un ejemplo extremo pero ilustra claramente el problema de estimar la probabilidad de supervivencia y transiciones en una población con un tamaño de muestra pequeña y puede resultar en estimados sesgados.
En las próximas secciones vemos métodos de simulaciones y estimaciones de intervalos de confianza para abordar la variación biológica en tiempo y espacio y la variación por el tamaño de muestra pequeña.
Tres fuentes de incertidumbre que las simulaciones pueden capturar.
Las tres pueden combinarse en un solo modelo, pero conviene evaluarlas por separado para entender cuál domina en una población dada.
Los métodos de simulaciones para estimar la variación en los parámetros tanto de la matriz como del crecimiento poblacional varían mucho. Lo que es consistente es que se utilizan datos disponibles para hacer simulaciones y estimar la variación en los parámetros. En otras palabras se usa la variación entre los elementos de múltiples matrices de transición en tiempo o espacio o el tamaño de muestra para hacer simulaciones y estimar la dispersión en los parámetros. La manera de observar la variación puede asumir diferentes formas, por ejemplo, la variación en los parámetros de la matriz de transición puede ser expresada como una distribución normal (distribución en forma de campana), una distribución de Poisson (datos con valores discretos, 0, 1, 2… etc.), una distribución binomial (distribución donde hay solamente dos alternativas) o beta (distribución donde el rango es limitado a los valores entre 0 y 1, inclusive), entre otras distribuciones. Reconocer el tipo de variación en los parámetros de la matriz de transición es importante para escoger el método de simulaciones y estimaciones de intervalos de confianza y reconocer sus limitaciones e interpretación.
En este caso vemos el efecto de la variación espacial en la probabilidad en los estimados. Aquí nos interesa descubrir y cuantificar cuán variables son las poblaciones unas de otras. El objetivo es determinar si las poblaciones se comportan de forma similar unas a otras o varían mucho de un sitio a otro.
El primer paso es definir las diferentes matrices, una por cada periodo de tiempo. En el siguiente script se enseña cómo entrar los datos para múltiples matrices en un solo objeto. Nota que es una lista de matrices.

Los datos provienen de un estudio de la dinámica de una población de Serapias cordigera de 1999 al 2012 evaluando el impacto antropogénico sobre la dinámica de la población (Pellegrino y Bellusci 2014). En este caso se emplean solamente 6 matrices de transición de la población de Serapias cordigera en 1999, tres de estas son poblaciones que tienen impacto antropogénico (A1, A2 y A3) y tres poblaciones naturales, sin impacto antropogénico (N1, N2, N3). Nota que se construye una lista (list()) de matrices de transición, una por cada periodo de tiempo.
Serapia <- list(
A1_1999 = matrix(
c(
0.745, 0.152, 0.452, 0.564,
0.125, 0.000, 0.000, 0.000,
0.000, 0.321, 0.205, 0.342,
0.200, 0.365, 0.205, 0.185
),
nrow = 4, byrow = TRUE,
dimnames = list(
c("latente", "plantula", "rosetta", "con flor"),
c("latente", "plantula", "rosetta", "con flor")
)
),
A2_1999 = matrix(
c(
0.648, 0.203, 0.414, 0.604,
0.188, 0.000, 0.000, 0.000,
0.000, 0.342, 0.198, 0.377,
0.242, 0.264, 0.225, 0.191
),
nrow = 4, byrow = TRUE,
dimnames = list(
c("latente", "plantula", "rosetta", "con flor"),
c("latente", "plantula", "rosetta", "con flor")
)
),
A3_1999 = matrix(
c(
0.544, 0.223, 0.364, 0.498,
0.148, 0.000, 0.000, 0.000,
0.000, 0.249, 0.243, 0.287,
0.242, 0.303, 0.210, 0.101
),
nrow = 4, byrow = TRUE,
dimnames = list(
c("latente", "plantula", "rosetta", "con flor"),
c("latente", "plantula", "rosetta", "con flor")
)
),
N1_1999 = matrix(
c(
0.287, 0.271, 0.054, 0.107,
0.318, 0.000, 0.000, 0.000,
0.000, 0.438, 0.228, 0.273,
0.111, 0.512, 0.585, 0.542
),
nrow = 4, byrow = TRUE,
dimnames = list(
c("latente", "plantula", "rosetta", "con flor"),
c("latente", "plantula", "rosetta", "con flor")
)
),
N2_1999 = matrix(
c(
0.299, 0.253, 0.066, 0.182,
0.324, 0.000, 0.000, 0.000,
0.000, 0.461, 0.222, 0.264,
0.339, 0.568, 0.554, 0.547
),
nrow = 4, byrow = TRUE,
dimnames = list(
c("latente", "plantula", "rosetta", "con flor"),
c("latente", "plantula", "rosetta", "con flor")
)
),
N3_1999 = matrix(
c(
0.245, 0.156, 0.052, 0.147,
0.325, 0.000, 0.000, 0.000,
0.000, 0.422, 0.305, 0.252,
0.210, 0.465, 0.498, 0.485
),
nrow = 4, byrow = TRUE,
dimnames = list(
c("latente", "plantula", "rosetta", "con flor"),
c("latente", "plantula", "rosetta", "con flor")
)
)
)
Serapia$A1_1999
latente plantula rosetta con flor
latente 0.745 0.152 0.452 0.564
plantula 0.125 0.000 0.000 0.000
rosetta 0.000 0.321 0.205 0.342
con flor 0.200 0.365 0.205 0.185
$A2_1999
latente plantula rosetta con flor
latente 0.648 0.203 0.414 0.604
plantula 0.188 0.000 0.000 0.000
rosetta 0.000 0.342 0.198 0.377
con flor 0.242 0.264 0.225 0.191
$A3_1999
latente plantula rosetta con flor
latente 0.544 0.223 0.364 0.498
plantula 0.148 0.000 0.000 0.000
rosetta 0.000 0.249 0.243 0.287
con flor 0.242 0.303 0.210 0.101
$N1_1999
latente plantula rosetta con flor
latente 0.287 0.271 0.054 0.107
plantula 0.318 0.000 0.000 0.000
rosetta 0.000 0.438 0.228 0.273
con flor 0.111 0.512 0.585 0.542
$N2_1999
latente plantula rosetta con flor
latente 0.299 0.253 0.066 0.182
plantula 0.324 0.000 0.000 0.000
rosetta 0.000 0.461 0.222 0.264
con flor 0.339 0.568 0.554 0.547
$N3_1999
latente plantula rosetta con flor
latente 0.245 0.156 0.052 0.147
plantula 0.325 0.000 0.000 0.000
rosetta 0.000 0.422 0.305 0.252
con flor 0.210 0.465 0.498 0.485Si uno asume que hay igual cantidad de poblaciones afectadas antropogénicamente y naturales, entonces se puede hacer un análisis de la variación espacial en la probabilidad de supervivencia. Se puede simular el crecimiento con igual probabilidad de selección de las matrices de transición. En otras palabras, cada matriz será seleccionada al azar 1/6 de las veces. Este supuesto es razonable si uno asume que hay igual cantidad de poblaciones afectadas antropogénicamente y naturales. El objetivo es ver la variación en la probabilidad de supervivencia en la población de Serapias cordigera en 1999, o más precisamente, cuál sería el tamaño poblacional típico de la especie si esta se comporta como las siguientes matrices (Pellegrino y Bellusci 2014).
La función stoch.projection() del paquete popbio permite hacer simulaciones de la dinámica de una población y da una lista de la cantidad de individuos esperado por cada etapa de la vida. Comenzamos por definir el tamaño de la población inicial por etapa de la vida, n, el nombre de las etapas de la vida y luego usamos la función stoch.projection() para hacer las simulaciones.
La función necesita tres argumentos, la lista de matrices de transición, el tamaño de la población inicial por etapa de la vida y el número de repeticiones. En este caso se usa 1000 repeticiones para tener una buena estimación de la variación en el tamaño de la población esperado por etapa de la vida. La función devuelve una lista con las simulaciones del tamaño de la población esperado por etapa de la vida.
n <- c(2, 0, 4, 99) # corresponde a la cantidad de individuos por etapa en la publicación del año 2000 del artículo Pellegrino et al. 2014
names(n) <- c("latente", "plantula", "rosetta", "con flor")
Serapia.eq <- stoch.projection(Serapia, n, nreps = 1000) # simulación con igual probabilidad de selección
as.data.frame(head(Serapia.eq)) |> flextable() # vemos las primeras 6 simulaciones de los tamaños de muestra esperada por etapalatente | plantula | rosetta | con flor |
|---|---|---|---|
2.007 | 0.1587 | 0.954 | 0.8822 |
5.019 | 0.3156 | 2.442 | 1.955 |
0.461 | 0.1982 | 0.619 | 1.158 |
0.3697 | 0.1422 | 0.6421 | 1.365 |
1.31 | 0.07427 | 0.625 | 0.6046 |
1.732 | 0.1203 | 0.8255 | 0.6874 |
Para observar cuántos individuos hubiese en el futuro de la población de Serapias cordigera si la especie se comporta como las matrices de transición. Se observa que las poblaciones tendrían pocos individuos, típicamente menos de 2-3 individuos en todas las etapas, sin importar si fuesen latente, plántula, rosetta o con flores.

Serapia.eq <- clean_names(as.data.frame(Serapia.eq)) # convertir la lista de matrices en un data frame y limpiar los nombres de las columnas
Serapia.eq %>%
gather(key = "etapa", value = "individuos") %>% # convertir el data frame de formato ancho a largo
mutate(etapa = factor(etapa, levels = c("plantula", "latente", "rosetta", "con_flor"))) %>% # ordenar las etapas de la vida
ggplot(aes(x = individuos)) +
geom_histogram(bins = 30, aes(fill = "blue"), color = "white") +
facet_wrap(~etapa, scales = "free") +
rlt_style_fill() +
labs(
title = "Distribución de número de individuos esperado de *Serapias cordigera*",
x = "Número de individuos", y = "Frecuencia"
) +
theme(title = element_markdown()) +
theme(axis.text.x = element_text(angle = 45, hjust = 1))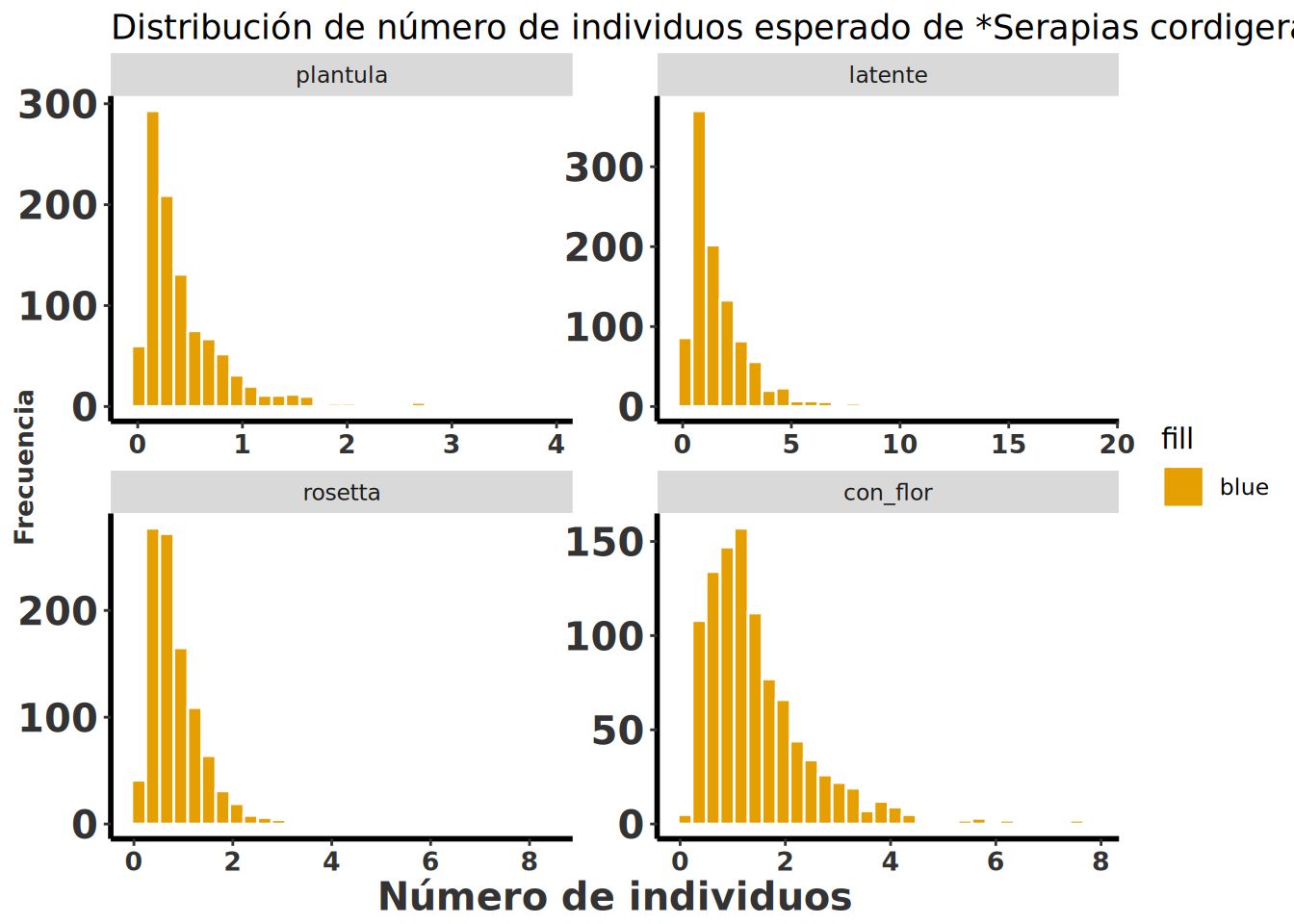
En la siguiente figura se suma el total de individuos por simulación, que es una medida de la cantidad de individuos esperada en el futuro. En el siguiente gráfico se muestra la distribución de la cantidad de individuos esperada en el futuro de la población de Serapias cordigera si la especie se comporta como las matrices de transición de arriba, seleccionadas al azar con la misma probabilidad. Se observa que la mayoría de las simulaciones tienen menos de 10 individuos en el futuro.
Serapia_eq_T <- Serapia.eq %>%
mutate(total = rowSums(across(where(is.numeric)))) # sumar la cantidad de individuos en una columna
ggplot(Serapia_eq_T, aes(x = total)) +
geom_histogram(bins = 30, aes(fill = "blue"), color = "white") +
rlt_style_fill() +
labs(
title = "Distribución de total de individuos esperado de *Serapias cordigera*",
x = "Número total de individuos por simulación", y = "Frecuencia"
) +
theme(title = element_markdown()) +
theme(legend.position = "none")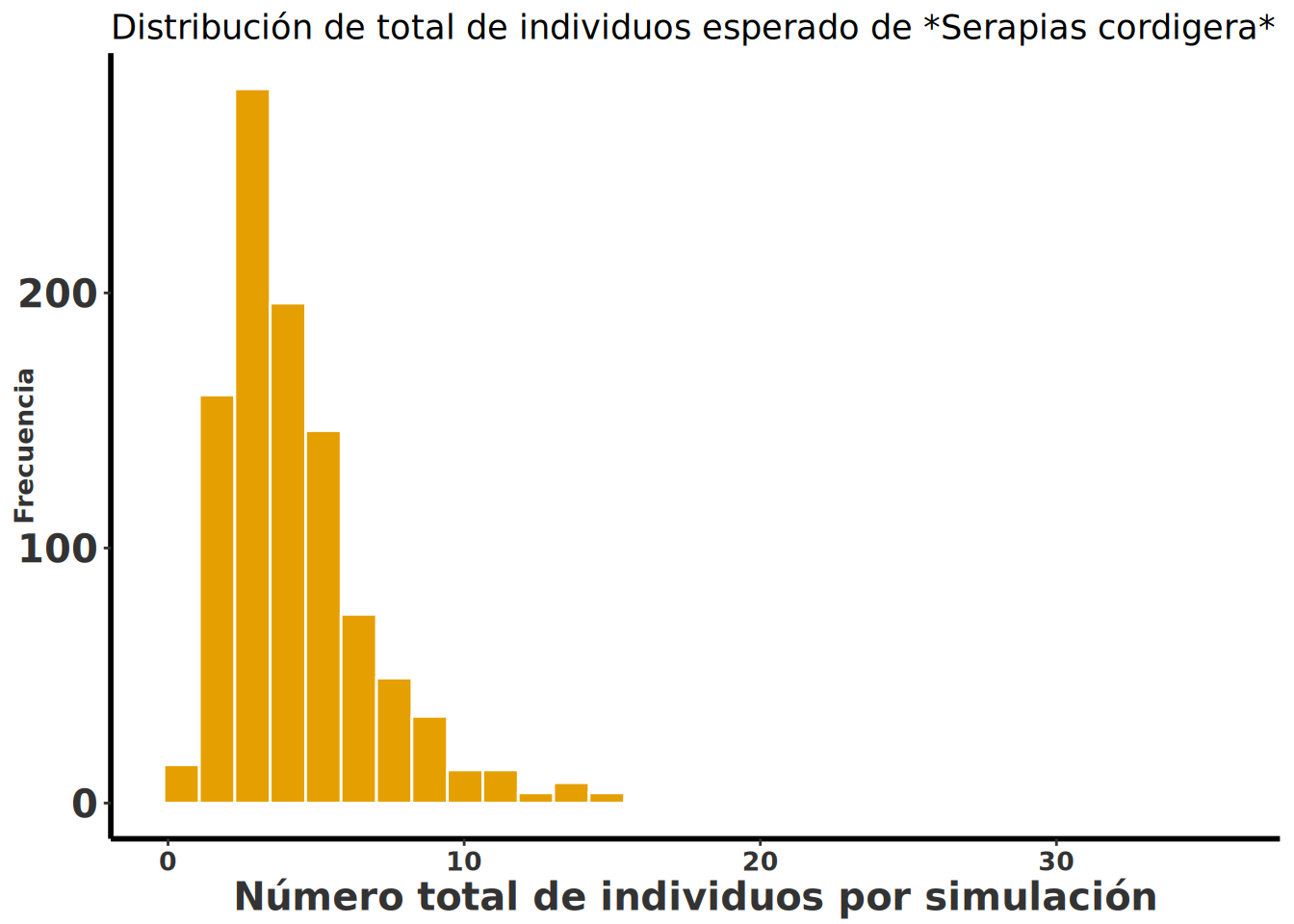
Las probabilidades de selección permiten modelar escenarios climáticos. Si entre las matrices disponibles algunas corresponden a años secos y otras a años típicos, asignar mayor probabilidad de selección a las matrices «secas» simula un escenario de mayor frecuencia de sequías —útil para evaluar respuestas poblacionales bajo cambio climático. Lo mismo aplica para huracanes, periodos de manejo intensivo, o cualquier evento esperado a frecuencia conocida.
La probabilidad de selección de las matrices de transición no tiene que ser igual. En el siguiente script se muestra cómo hacer simulaciones con diferente probabilidad de selección de las matrices de transición. En este caso se asume que las 3 poblaciones afectadas antropogénicamente son más comunes que las poblaciones naturales. Las 3 poblaciones naturales son raras y poco comunes. En el script vemos que ahora las poblaciones afectadas antropogénicamente se seleccionan con una probabilidad de 0.85 (0.30 + 0.30 + 0.25) y las poblaciones naturales con una probabilidad de 0.15. En otras palabras, las poblaciones naturales son muy raras y la especie está dominada por sitios donde se ve afectada antropogénicamente. Importante: esta es una simulación y no refleja la realidad de la especie ni de la publicación.
n <- c(2, 0, 4, 99) # población inicial por etapa
names(n) <- c("latente", "plantula", "rosetta", "con flor")
Serapia.uneq <- stoch.projection(Serapia, n, nreps = 1000, prob = c(.3, .3, .25, .05, .05, .05)) # simulación con igual probabilidad de selección
as.data.frame(head(Serapia.uneq)) |> flextable() # vemos las primeras 6 simulaciones de tamaño de muestralatente | plantula | rosetta | con flor |
|---|---|---|---|
8.998 | 1.769 | 1.899 | 3.58 |
23.74 | 4.363 | 5.044 | 10.36 |
28.47 | 5.171 | 7.274 | 11.67 |
25.94 | 2.894 | 5.843 | 9.166 |
13.75 | 2.677 | 2.936 | 5.472 |
22.39 | 2.254 | 5.139 | 7.546 |
Ahora vamos a solapar las dos distribuciones (la de selección por igual y por poblaciones mayormente impactado por humanos) para ver si hay una diferencia en la distribución de tamaño de muestra entre las dos simulaciones. Los resultados de las simulaciones sugieren que las poblaciones dominadas por impacto antropogénico (naranja) tienen una mayor cantidad de individuos en el futuro que las poblaciones bajo igualdad de probabilidades (gris).
Serapia.uneq <- clean_names(as.data.frame(Serapia.uneq))
Serapia.uneq_T <- Serapia.uneq %>%
mutate(total = rowSums(across(where(is.numeric))))
ggplot(Serapia.uneq_T, aes(total)) +
geom_histogram(bins = 30, aes(fill = "blue"), color = "white") +
geom_histogram(data = Serapia_eq_T, aes(x = total), bins = 30,colour="black", alpha = .7) +
rlt_style_fill() +
labs(
title = "Distribución del total de individuos esperado de *Serapias cordigera*",
x = "Número total de individuos por simulación", y = "Frecuencia"
) +
theme(title = element_markdown()) +
theme(legend.position = "none")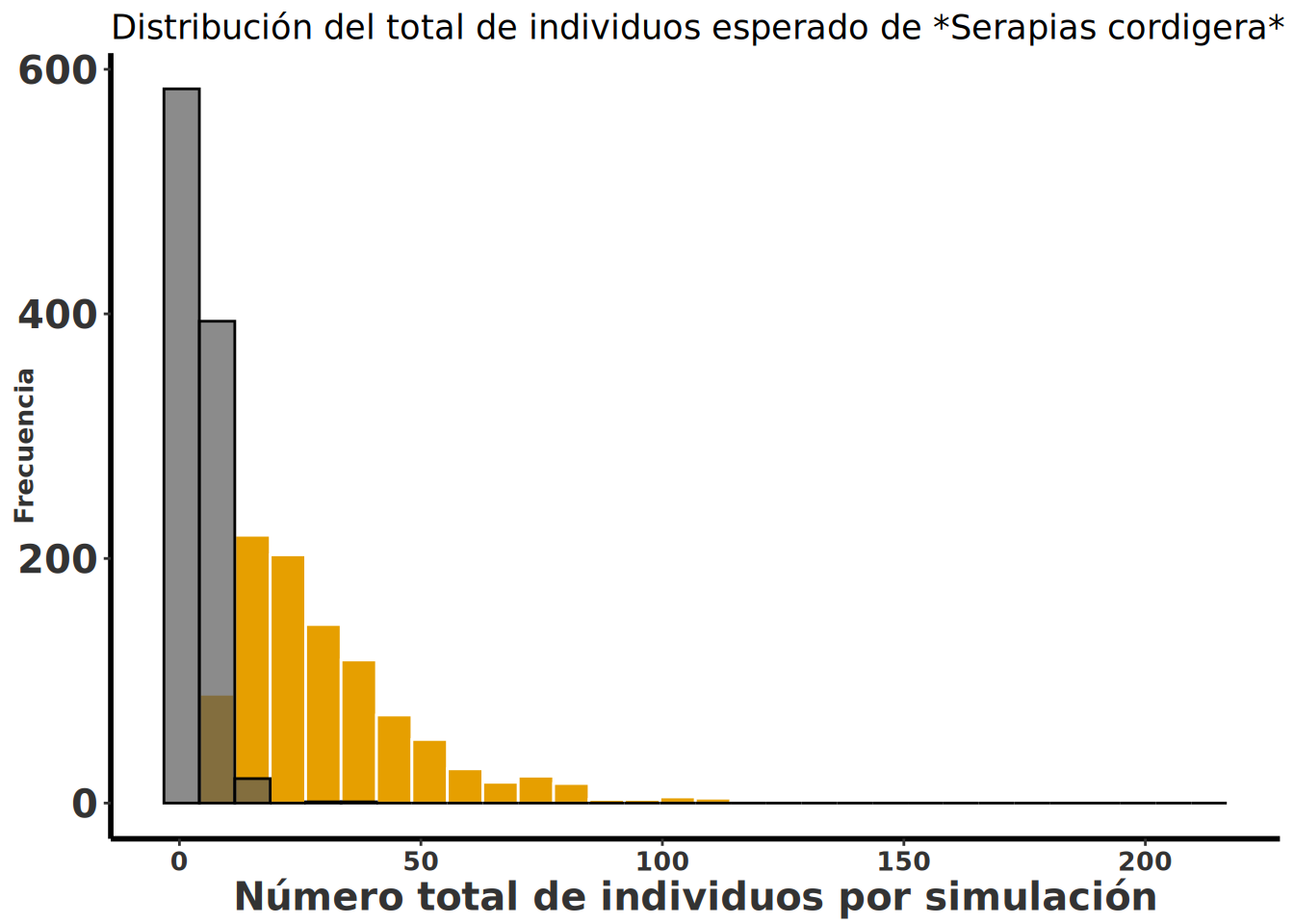
En el siguiente script podemos evaluar el crecimiento poblacional y su variación en el espacio.
Nota: aquí se calcula el log del crecimiento poblacional estocástico de Serapias cordigera por aproximación y simulaciones siguiendo la fórmula de Tuljapurkar (Tuljapurkar 2013). Se puede ver que la especie tiene un crecimiento poblacional negativo en la mayoría de las simulaciones. Esto sugiere que la especie está en declive. Pero también se puede ver que hay variación en el crecimiento poblacional de la especie. El script incluye un parámetro para indicar por cuánto tiempo queremos que la simulación ocurra, maxt=; en este caso se simula por 50 periodos de tiempo. Si no se añade nada, la simulación dura por 50000 periodos de tiempo. Nota que maxt es en la unidad de tiempo de la matriz de transición; si esta fue construida en un periodo de meses, entonces la simulación opera en ese intervalo de tiempo.
Cuando se usa el log de crecimiento poblacional estocástico se calcula \(r\), donde \(r = \ln(\lambda)\). La interpretación de \(r\) es distinta a la de \(\lambda\):
El crecimiento estocástico de las poblaciones de Serapias cordigera en 1999 es negativo, lo que sugiere que la especie está en declive. Pero también se puede ver que hay variación en el crecimiento poblacional de la especie. Se usa la función stoch.growth.rate().
Simulaciones por selección de matriz con igual probabilidad
SGR_eq <- stoch.growth.rate(Serapia, maxt = 50, verbose = TRUE) # matrices seleccionado con igual probabilidad
SGR_eq$approx
[1] -0.06760619
$sim
[1] -0.03933639
$sim.CI
[1] -0.075606717 -0.003066061SGR_uneq <- stoch.growth.rate(Serapia, maxt = 50, prob = c(.3, .3, .25, .05, .05, .05), verbose = FALSE) # matrices seleccionado con diferente probabilidades
SGR_uneq$approx
[1] -0.02988694
$sim
[1] -0.03130341
$sim.CI
[1] -0.060052923 -0.002553901Para convertir el log(crecimiento poblacional estocástico) a lambda se usa la siguiente fórmula \(e^r\) donde \(r\) es el log(crecimiento poblacional estocástico). Como ejemplo se muestra la metodología con las simulaciones con probabilidad desigual de selección de las matrices de transición.
Por qué stoch.growth.rate() reporta \(\log \lambda_s\) y no \(\lambda_s\) directamente. Cuando una población crece estocásticamente, la media geométrica del crecimiento (no la aritmética) es el predictor correcto del tamaño futuro. La media geométrica de \(\lambda\) se calcula como \(\exp(\overline{\log \lambda})\). Por eso conviene trabajar en escala logarítmica internamente y convertir a \(\lambda\) con exp() solo al reportar. Si los intervalos de confianza incluyen 0 en la escala log, son equivalentes a incluir 1 en la escala \(\lambda\) — es decir, no hay evidencia de crecimiento ni declive.
Lambda — probabilidad desigual de selección
exp(SGR_uneq$sim) # matrices seleccionadas con probabilidad desigual[1] 0.9691815exp(SGR_uneq$sim.CI[1]) # El intervalo de confianza inferior[1] 0.9417147exp(SGR_uneq$sim.CI[2]) # El intervalo de confianza superior[1] 0.9974494Los análisis anteriores no separan las matrices de transición por el impacto antropogénico versus las poblaciones naturales. En el siguiente script se muestra cómo hacer simulaciones y estimar el crecimiento poblacional de las poblaciones afectadas, de forma separada.
Serapia_A <- Serapia[1:3] # poblaciones afectadas antropogénicamente
Serapia_N <- Serapia[4:6] # poblaciones naturalesAsumimos una probabilidad igual de selección por cada simulaciones en ambos escenarios
n <- c(2, 0, 4, 99) # corresponde a los datos de la especie en la publicación del año 2000
names(n) <- c("latente", "plantula", "rosetta", "con flor")
Serapia.eq_A <- stoch.projection(Serapia_A, n, nreps = 1000)
Serapia.eq_N <- stoch.projection(Serapia_N, n, nreps = 1000)Convertir las simulaciones de las matrices de transición en un data frame y limpiar los nombres de las columnas. Luego sumar el total de individuos por simulación para cada etapa de la vida. Esto es necesario para poder visualizar el tamaño poblacional esperado por etapa de la vida. El resultado del análisis es que las poblaciones afectadas antropogénicamente tienen un estimado de tamaño poblacional más grande que las poblaciones naturales.
Serapia.eq_A <- clean_names(as.data.frame(Serapia.eq_A)) # limpiar los nombres de las columnas y convertir en un data frame
Serapia.eq_N <- clean_names(as.data.frame(Serapia.eq_N))
Serapia_eq_A <- Serapia.eq_A %>%
mutate(total = rowSums(across(where(is.numeric)))) # sumar la cantidad de individuos en una columna
Serapia_eq_N <- Serapia.eq_N %>%
mutate(total = rowSums(across(where(is.numeric))))Visualizar el tamaño poblacional esperado por etapa de la vida para las poblaciones afectadas antropogénicamente y naturales. Se observa que las poblaciones afectadas antropogénicamente tienen un tamaño poblacional más grande que las poblaciones naturales; las poblaciones con efecto antropogénico tienden a ser de mayor cantidad.
diverging <- c("#E69F00", "#56B4E9", "#009392", "#F0E442", "#0072B2", "#D55E00", "#CC79A7", "#000000")
ggplot(Serapia_eq_N, aes(total)) +
geom_histogram(fill = "#009392", color = "white", bins = 30, alpha = .5) +
geom_histogram(
data = Serapia_eq_A, aes(x = total),
fill = "#CF597E", color = "white", bins = 30, alpha = .5
) +
theme_minimal() +
labs(
title = "Distribución de total de individuos esperado de *Serapias cordigera*",
x = "Número total de individuos por simulación", y = "Frecuencia"
) +
theme(title = element_markdown())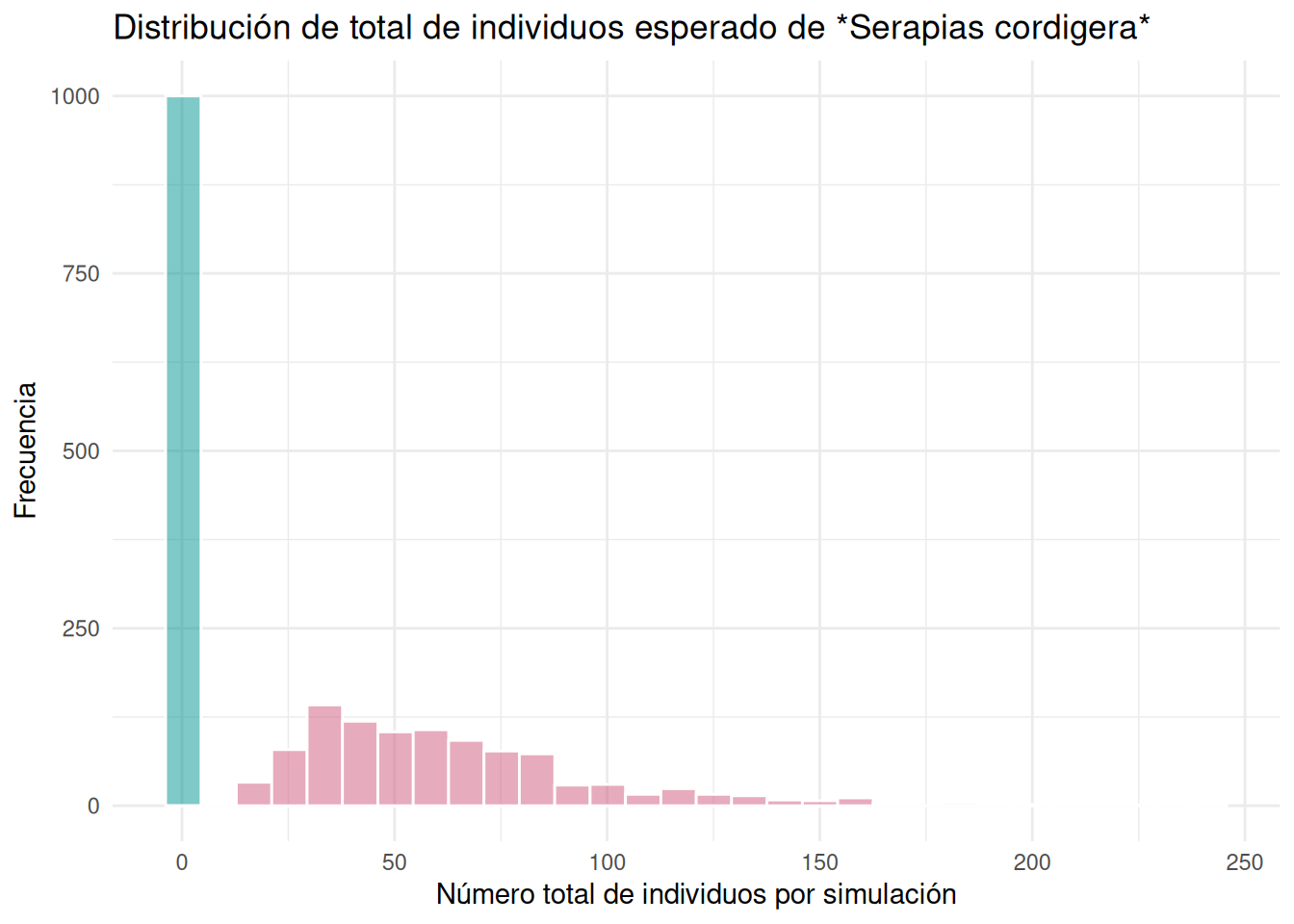

El objetivo de evaluar la variación en el tiempo es determinar si los efectos temporales son importantes para predecir los cambios en el crecimiento o la estabilidad poblacional. Seguimos con los datos de Serapias cordigera, pero ahora evaluamos cuál es el efecto de la variación en los parámetros de la matriz usando múltiples censos. Vemos el efecto en una de esas poblaciones con impacto antropogénico, solamente la población A1, en los 13 años de muestreo, desde 1999 hasta 2011, inclusive.
El primer paso es entrar los datos en una lista de matrices de transición.
Serapia_tiempo <- list(
A1_1999 = matrix(
c(
0.745, 0.152, 0.452, 0.564,
0.125, 0.000, 0.000, 0.000,
0.000, 0.321, 0.205, 0.342,
0.200, 0.365, 0.205, 0.185
),
nrow = 4, byrow = TRUE,
dimnames = list(
c("latente", "plantula", "rosetta", "con flor"),
c("latente", "plantula", "rosetta", "con flor")
)
),
A1_2000 = matrix(
c(
0.578, 0.222, 0.394, 0.394,
0.188, 0.000, 0.000, 0.000,
0.000, 0.322, 0.254, 0.357,
0.244, 0.314, 0.125, 0.302
),
nrow = 4, byrow = TRUE,
dimnames = list(
c("latente", "plantula", "rosetta", "con flor"),
c("latente", "plantula", "rosetta", "con flor")
)
),
A1_2001 = matrix(
c(
0.668, 0.122, 0.294, 0.401,
0.128, 0.000, 0.000, 0.000,
0.000, 0.302, 0.453, 0.366,
0.214, 0.364, 0.185, 0.207
),
nrow = 4, byrow = TRUE,
dimnames = list(
c("latente", "plantula", "rosetta", "con flor"),
c("latente", "plantula", "rosetta", "con flor")
)
),
A1_2002 = matrix(
c(
0.645, 0.222, 0.682, 0.504,
0.177, 0.000, 0.000, 0.000,
0.000, 0.421, 0.305, 0.382,
0.102, 0.355, 0.208, 0.208
),
nrow = 4, byrow = TRUE,
dimnames = list(
c("latente", "plantula", "rosetta", "con flor"),
c("latente", "plantula", "rosetta", "con flor")
)
),
A1_2003 = matrix(
c(
0.545, 0.113, 0.424, 0.424,
0.247, 0.000, 0.000, 0.000,
0.000, 0.166, 0.233, 0.277,
0.192, 0.253, 0.209, 0.212
),
nrow = 4, byrow = TRUE,
dimnames = list(
c("latente", "plantula", "rosetta", "con flor"),
c("latente", "plantula", "rosetta", "con flor")
)
),
A1_2004 = matrix(
c(
0.548, 0.191, 0.402, 0.527,
0.327, 0.000, 0.000, 0.000,
0.000, 0.338, 0.127, 0.394,
0.211, 0.262, 0.296, 0.142
),
nrow = 4, byrow = TRUE,
dimnames = list(
c("latente", "plantula", "rosetta", "con flor"),
c("latente", "plantula", "rosetta", "con flor")
)
),
A1_2005 = matrix(
c(
0.602, 0.153, 0.486, 0.562,
0.224, 0.000, 0.000, 0.000,
0.000, 0.491, 0.324, 0.304,
0.139, 0.268, 0.294, 0.277
),
nrow = 4, byrow = TRUE,
dimnames = list(
c("latente", "plantula", "rosetta", "con flor"),
c("latente", "plantula", "rosetta", "con flor")
)
),
A1_2006 = matrix(
c(
0.487, 0.471, 0.425, 0.607,
0.318, 0.000, 0.000, 0.000,
0.000, 0.438, 0.328, 0.373,
0.211, 0.212, 0.285, 0.142
),
nrow = 4, byrow = TRUE,
dimnames = list(
c("latente", "plantula", "rosetta", "con flor"),
c("latente", "plantula", "rosetta", "con flor")
)
),
A1_2007 = matrix(
c(
0.544, 0.423, 0.374, 0.578,
0.348, 0.000, 0.000, 0.000,
0.000, 0.441, 0.248, 0.288,
0.145, 0.203, 0.311, 0.201
),
nrow = 4, byrow = TRUE,
dimnames = list(
c("latente", "plantula", "rosetta", "con flor"),
c("latente", "plantula", "rosetta", "con flor")
)
),
A1_2008 = matrix(
c(
0.568, 0.325, 0.294, 0.607,
0.227, 0.000, 0.000, 0.000,
0.000, 0.402, 0.553, 0.256,
0.194, 0.364, 0.275, 0.217
),
nrow = 4, byrow = TRUE,
dimnames = list(
c("latente", "plantula", "rosetta", "con flor"),
c("latente", "plantula", "rosetta", "con flor")
)
),
A1_2009 = matrix(
c(
0.545, 0.356, 0.472, 0.398,
0.225, 0.000, 0.000, 0.000,
0.000, 0.322, 0.304, 0.452,
0.110, 0.225, 0.298, 0.185
),
nrow = 4, byrow = TRUE,
dimnames = list(
c("latente", "plantula", "rosetta", "con flor"),
c("latente", "plantula", "rosetta", "con flor")
)
),
A1_2010 = matrix(
c(
0.399, 0.352, 0.486, 0.580,
0.424, 0.000, 0.000, 0.000,
0.000, 0.561, 0.312, 0.264,
0.239, 0.168, 0.354, 0.247
),
nrow = 4, byrow = TRUE,
dimnames = list(
c("latente", "plantula", "rosetta", "con flor"),
c("latente", "plantula", "rosetta", "con flor")
)
),
A1_2011 = matrix(
c(
0.497, 0.352, 0.465, 0.622,
0.324, 0.000, 0.000, 0.000,
0.000, 0.361, 0.321, 0.264,
0.139, 0.368, 0.367, 0.177
),
nrow = 4, byrow = TRUE,
dimnames = list(
c("latente", "plantula", "rosetta", "con flor"),
c("latente", "plantula", "rosetta", "con flor")
)
)
)
# Serapia_tiempoEl análisis evalúa la variación temporal en el crecimiento poblacional de la especie de Serapias cordigera en 1999 a 2012. Usando la función stoch.projection() se observa la variación en cantidad de individuos por etapa.
n <- c(2, 0, 4, 99) # población inicial por etapa
names(n) <- c("latente", "plantula", "rosetta", "con flor")
Serapia_tiempo_eq <- stoch.projection(Serapia_tiempo, n, nreps = 5000) # simulación con igual probabilidad de selección
as.data.frame(head(Serapia_tiempo_eq)) |> flextable() # vemos las primeras 6 simulaciones de tamaño de muestralatente | plantula | rosetta | con flor |
|---|---|---|---|
63.41 | 15.02 | 24.5 | 22.53 |
44.08 | 10.66 | 17.16 | 15.37 |
115.5 | 18.14 | 38.05 | 35.73 |
56.75 | 13.13 | 15.15 | 20.79 |
82.75 | 39.22 | 35.88 | 46.68 |
63.55 | 19.03 | 24.32 | 25.97 |
Para evaluar el cambio de crecimiento poblacional en la especie de Serapias cordigera en 1999 a 2011 se usa la función stoch.growth.rate().
Se observa que el crecimiento poblacional es cerca de cero en la mayoría de las simulaciones, lo que sugiere que la especie no está en declive ni creciendo. Los intervalos de confianza solapan el cero, dando apoyo a que el crecimiento temporal de la población no es diferente del de una población estable durante este periodo de tiempo.
Nota que se puede cambiar la probabilidades de selección de las matrices de transición, en este caso se asume que hay igual cantidad de poblaciones afectadas antropogénicamente y naturales. Pero considera por ejemplo que hay eventos abióticos como temperatura o lluvia fuera de lo común en un año, entonces se puede cambiar la probabilidad de selección de las matrices de transición para evaluar el efecto de estos eventos de cambio climatológico en el crecimiento poblacional de la especie. Si, por ejemplo, en el área donde están estas especies hay predicción de que un factor abiótico —por ejemplo, una reducción en la lluvia (aumento en la sequía)— cambiará en las próximas décadas, se pudiese cambiar la probabilidad de selección de las matrices que corresponden a periodos de sequía para que sean seleccionadas más frecuentemente en las simulaciones. El objetivo es de simular los efectos de cambio en tiempo sobre el crecimiento poblacional de la especie basado en un patrón esperado de factores abióticos o bióticos.
SGR_Temp_eq <- stoch.growth.rate(Serapia_tiempo, maxt = 50) # matrices seleccionado con igual probabilidad por 50 años
SGR_Temp_eq$approx
[1] 0.002712947
$sim
[1] 0.02127045
$sim.CI
[1] -0.002618836 0.045159734# Conversión a valores de Lambda
exp(SGR_Temp_eq$sim) # Lambda de matrices seleccionado con igual probabilidad[1] 1.021498exp(SGR_Temp_eq$sim.CI[1]) # Lambda del intervalo de confianza inferior[1] 0.9973846exp(SGR_Temp_eq$sim.CI[2]) # Lambda del intervalo de confianza superior[1] 1.046195Comparar la variación temporal en el crecimiento poblacional de la especie de Serapias cordigera en 1999 a 2011 con la variación espacial.
Lo que se observa es que la variación temporal y la variación espacial es más o menos igual (el rango de variación, diff), pero que hay menos variación temporal que espacial. Por lo menos para los datos seleccionado de (Pellegrino y Bellusci 2014) la variación temporal no es tan grande como la variación espacial y solapa el lambda de 1, al contrario la variación espacial no solapa el lambda de 1 (ambos CI son menores de 1, sugiriendo una reducción posible de las poblaciones). Note que no usamos todos los datos de Pellegrino (Pellegrino y Bellusci 2014) pero un sub-grupo y por consecuencia el análisis es sesgado.
tipo | lambda | CI_inf | CI_sup | diff_ci |
|---|---|---|---|---|
Espacial | 0.9614 | 0.9272 | 0.9969 | 0.06976 |
Temporal | 1.021 | 0.9974 | 1.046 | 0.04881 |
Observación de la variación temporal y espacial en el crecimiento poblacional de la especie de Serapias cordigera en un gráfico.
Se nota que el crecimiento poblacional intrínseco de Serapias cordigera es negativo en la mayoría de las simulaciones cuando se evalúan múltiples poblaciones, pero la población A1, en general, tiene un crecimiento positivo o es estable cuando se evalúa tomando en cuenta la variación temporal. La línea horizontal representa un crecimiento poblacional de 1; si el crecimiento poblacional es mayor de 1, la población crece; si es menor de 1, la población decrece. En el análisis temporal vemos que el intervalo de confianza solapa el 1, lo que sugiere que no hay evidencia de que el crecimiento poblacional sea diferente del de una población estable.
ggplot(Tabla_resultado, aes(x = tipo, y = lambda, color = tipo)) +
geom_point(stat = "identity", colour = "black") +
geom_errorbar(aes(ymin = CI_inf, ymax = CI_sup), width = .2) +
rlt_style_colour() +
geom_hline(yintercept = 1, linetype = "dashed", color = "red") +
labs(title = "Comparación de simulaciones de crecimiento poblacional de *Serapias cordigera*") +
labs(x = "Tipo de variación", y = "Crecimiento poblacional") +
theme(title = element_markdown()) +
scale_y_continuous(limits = c(0.90, 1.1))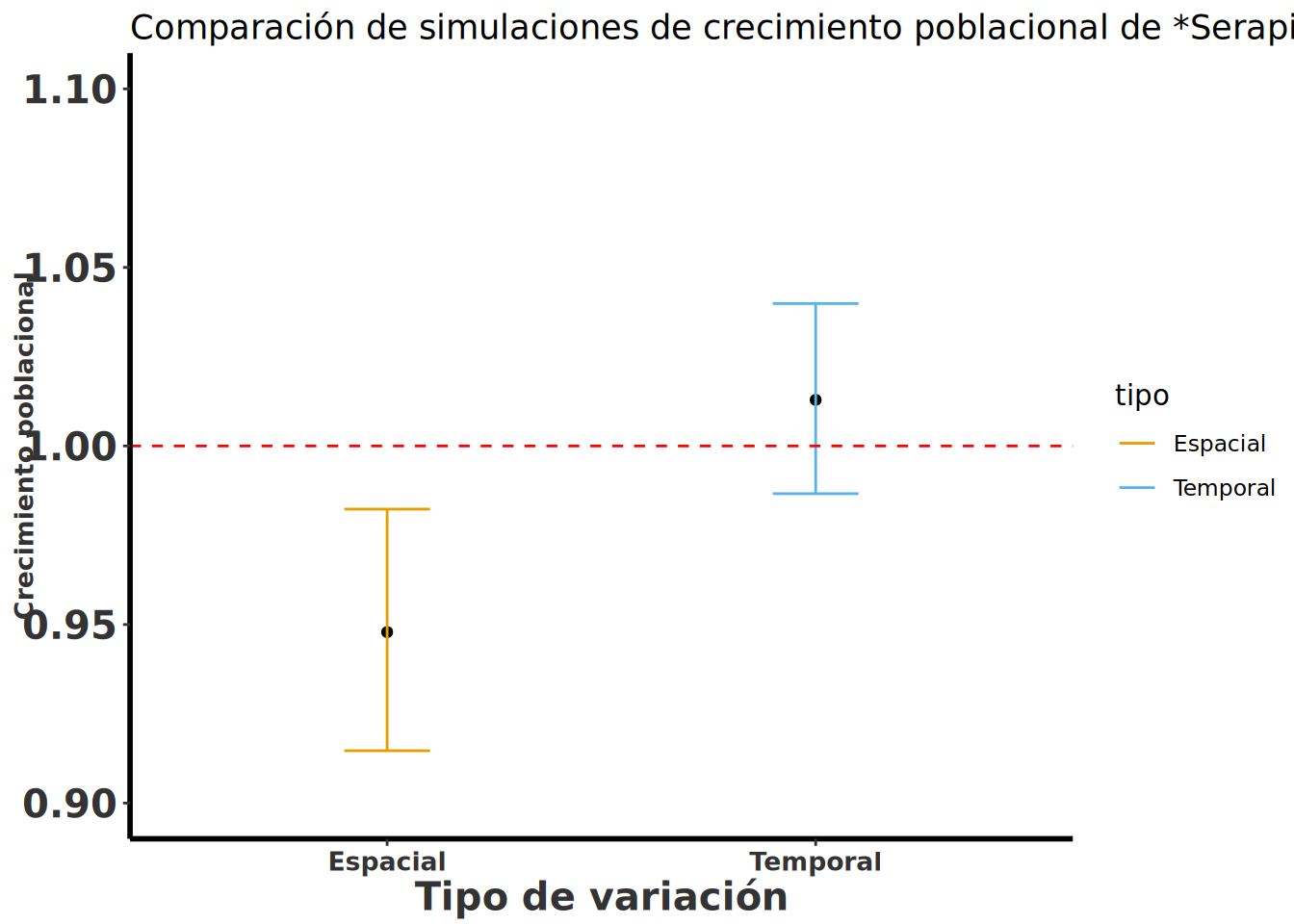
En esta sección vemos el efecto del tamaño de muestra sobre los parámetros de la matriz de transición.
Este método usa un acercamiento bayesiano y una distribución de probabilidad beta para estimar los parámetros de la matriz de transición conocida como la distribución Dirichlet múltiples para grupos discretos. Primero vemos a que se refiere un análisis bayesiano y la función Dirichlet. En lo que corresponde al presente análisis bayesiano, incluimos una previa al análisis; en otras palabras, se usa información previa para incluirla en los cálculos al estimar los parámetros, que se llaman los estimados posteriores. En nuestro caso haremos una previa con poco peso, en otra palabra, no influirá mucho (o casi nada) en los resultados. El concepto de previa es importante en el análisis bayesiano, es inherente a la filosofía de la estadística bayesiana. Se asume que la información anterior al estudio ayuda a tomar decisiones en los cálculos con los datos actuales. Como biólogos, siempre tenemos información previa; por ejemplo, sabemos que la probabilidad de que un individuo muera no es cero, y aunque esto es evidente, es una información adquirida por nuestra experiencia previa. Ahora, cuál es la probabilidad exacta de la muerte de un individuo es lo que se estima en el análisis bayesiano, tomando en cuenta tanto la información previa como la información recolectada de un estudio.
La función Dirichlet es una expansión de una función bien básica de probabilidad, usando una distribución beta. La función beta es una distribución de probabilidad continua en el intervalo de [0,1] y se usa para modelar la probabilidad de un evento binario. La función Dirichlet es una generalización de la función beta y se usa para modelar la probabilidad de un evento multinomial (múltiples grupos) donde la suma de cada probabilidades es igual a 1 y las probabilidades tienen que estar dentro del intervalo de [0,1]. En otra palabra no puede haber valores menores de 0 o mayores de 1, incluyendo la suma de estos.
Comenzamos con lo más básico, probabilidades de dos grupos. Consideramos un grupo de 10 individuos, si 6 de estos individuos sobreviven, cuál es la proporción que no sobrevive. La probabilidad de sobrevivir es \(p=6/10\), entonces la probabilidad de no sobrevivir es \(\hat{p}=1-p\), porque siempre la suma de \(p + \hat{p}= 1\).
Si tenemos tres grupos, digamos, plántulas que se quedan como plántula, que crecen a juveniles o que mueren. En este caso tenemos tres grupos (multinomial) y la probabilidad de que un individuo de las proporciones tiene que sumar a 1. En este caso la probabilidad de que un individuo se queda como plántula es \(p_1=0.5\), la probabilidad de que un individuo crece a juvenil es \(p_2=0.3\) y la probabilidad de que un individuo muere es \(p_3=0.2\). La suma de estas probabilidades tiene que ser igual a 1, en otra palabra \(p_1 + p_2 + p_3 = 1\).
La otra ventaja de usar la función Dirichlet es que se basa en la distribución beta. Esta es una distribución donde tanto los parámetros como el promedio y los intervalos de confianza están limitados a 0 y 1. Otra manera de decir lo mismo es que no se pueden tener intervalos de confianza menores que 0 o mayores que 1. Hagan el ejercicio mental de tratar de entender un intervalo de confianza con una probabilidad de -0.10. Nota que si uno usa una distribución normal, los intervalos de confianza pueden ser negativos o mayores de 1, lo que no tiene sentido biológico ni matemático en el contexto de probabilidad. De allí viene la ventaja de usar la distribución beta para calcular los parámetros y tener claro qué distribución se usa para estimar los parámetros.
Se selecciona la población A3 de Serapias cordigera para hacer el análisis. Esta población es una de las poblaciones afectadas antropogénicamente, y tiene un tamaño de muestra pequeño, lo que hace que sea interesante para ver el efecto del tamaño de muestra en los parámetros de la matriz de transición.
n <- c(2, 0, 4, 99) # población inicial por etapa
names(n) <- c("latente", "plantula", "rosetta", "con flor")
Serapia_A3 <- splitA(Serapia$A3_1999) # separar la matriz A3 en sus componentes (transición y fecundidad)En el siguiente paso vamos a asignar una previa a los datos. Estas previas provienen de Tremblay, no representan la visión de los autores del artículo. Se añade una previa también para la proporción de plantas que fallece en cada etapa (la quinta fila). Nota que es importante que la suma de cada columna sea igual a 1. Típicamente uno debería obtener la previa de la literatura o de un especialista en la biología de la especie antes de la recolección de datos, pero en este caso no se tiene información previa.
RLT_Serapia_previa <- matrix(
c(
0.5, 0.1, 0.4, 0.4,
0.2, 0., 0.0, 0.0,
0.0, 0.025, 0.25, 0.3,
0.15, 0.05, 0.25, 0.2,
0.05, 0.025, 0.1, 0.1
),
byrow = TRUE, nrow = 5, ncol = 4
)La distribución posterior marginal de un elemento en un multinomio es una distribución beta, y usamos esto para obtener intervalos creíbles en nuestras tasas de transición. Podemos usar el tipo de retorno TN para obtener los parámetros del multinomio deseado.
Nota que:
En el script vemos los intervalos de confianza de la matriz de transición de Serapias cordigera en 1999 para la población A3.
Lo más dramático de ese primer análisis es que los intervalos de credibilidad son muy grandes: casi todos los intervalos de credibilidad lcl y ucl van de muy cerca de cero a valores muy grandes. Esto sugiere que hay mucha incertidumbre en los datos. Este resultado es esperado, porque la muestra es muy pequeña; solamente 2 individuos fueron muestreados en el campo de esta etapa.
TN <- fill_transitions(Serapia_A3, N = n, P = RLT_Serapia_previa, priorweight = .01, returnType = "TN")
a <- TN[, 1] # cambie 1 a 2, 3 etc para obtener la distribución beta marginal de cada columna, el valor representa la columna.
b <- sum(TN[, 1]) - TN[, 1] # cambie 1 a 2, 3 etc para obtener la distribución beta marginal de cada columna.
p <- a / (a + b)
lcl <- qbeta(0.025, a, b)
ucl <- qbeta(0.975, a, b)
A3 <- knitr::kable(sprintf("%.3f, %.3f, %.3f", p, lcl, ucl))
A3df_lat <- round(data.frame(p, lcl, ucl), digits = 3)
A3df_lat$etapa_futura <- c("latente", "plantula", "rosetta", "con flor", "fallece")
A3df_lat p lcl ucl etapa_futura
1 0.544 0.038 0.984 latente
2 0.149 0.000 0.735 plantula
3 0.000 0.000 0.000 rosetta
4 0.241 0.000 0.844 con flor
5 0.066 0.000 0.544 falleceAhora vemos que pasa con la etapa que tenemos más datos, los adultos con flores. En este caso vemos que los intervalos de credibilidad son más pequeños, pero todavía son grandes, ya que el tamaño de muestra es de 99 para esta etapa.
TN <- fill_transitions(Serapia_A3, N = n, P = RLT_Serapia_previa, priorweight = 0.01, returnType = "TN")
a <- TN[, 4] # cambie 1 a 2, 3 etc para obter la distribución beta marginal de cada columna.
b <- sum(TN[, 4]) - TN[, 4] # cambie 1 a 2, 3 etc para obter la distribución beta marginal de cada columna.
p <- a / (a + b)
lcl <- qbeta(0.025, a, b)
ucl <- qbeta(0.975, a, b)
A3 <- knitr::kable(sprintf("%.3f, %.3f, %.3f", p, lcl, ucl))
A3df <- round(data.frame(p, lcl, ucl), digits = 3)
A3df$etapa_futura <- c("latente", "plantula", "rosetta", "con flor", "fallece")
A3df p lcl ucl etapa_futura
1 0.004 0.000 0.022 latente
2 0.000 0.000 0.000 plantula
3 0.287 0.203 0.379 rosetta
4 0.102 0.051 0.168 con flor
5 0.607 0.510 0.700 fallece
La simulación no inventa información que no está en los datos. Una población con sólo 2 plántulas observadas tendrá intervalos de credibilidad muy amplios, y eso es lo correcto: refleja la verdadera incertidumbre del muestreo. Aumentar el número de simulaciones (nreps) no estrecha los intervalos — sólo los estima con más precisión. Para estrechar intervalos se necesitan más datos de campo (más individuos marcados, más años de muestreo) o previas informativas más fuertes (justificadas por conocimiento experto). El enfoque bayesiano hace explícito este tradeoff entre datos y conocimiento previo.
Como ejercicio, vemos qué tipo de intervalos tendríamos si tuviésemos un tamaño de muestra de 500 individuos en la etapa de adulto. Nota que, aunque los intervalos de credibilidad son más pequeños, no se debería asumir que no hay variación demográfica en la etapa de adulto, por procesos estocásticos.
n500 <- c(2, 0, 4, 500) # población inicial por etapa
TN <- fill_transitions(Serapia_A3, N = n500, P = RLT_Serapia_previa, priorweight = 0.01, returnType = "TN")
a500 <- TN[, 4] # cambie 1 a 2, 3 o 4 etc. para obtener la distribución beta marginal de cada columna (etapas).
b500 <- sum(TN[, 4]) - TN[, 4] # cambie 1 a 2, 3 etc. para obtener la distribución beta marginal de cada columna.
p500 <- a500 / (a500 + b500)
lcl <- qbeta(0.025, a500, b500)
ucl <- qbeta(0.975, a500, b500)
A3 <- knitr::kable(sprintf("%.3f, %.3f, %.3f", p, lcl, ucl))
A3df <- round(data.frame(p, lcl, ucl), digits = 3)
A3df$etapa_futura <- c("latente", "plantula", "rosetta", "con flor", "fallece")
A3df p lcl ucl etapa_futura
1 0.004 0.000 0.011 latente
2 0.000 0.000 0.000 plantula
3 0.287 0.249 0.327 rosetta
4 0.102 0.077 0.130 con flor
5 0.607 0.564 0.649 falleceAhora vemos cómo se distribuye una distribución beta. Usamos los datos de la etapa de adulto con flores con 99 individuos. Se observa que ninguna de las transiciones o estasis tiene una distribución normal. La distribución que demuestra bien este patrón es la de los adultos con flores que pasan a latente. La línea roja vertical representa el promedio, es decir, el valor en la matriz de transición.
a # los valores del shape alpha[1] 0.396 0.000 28.710 10.197 60.687b # los valores del shape beta[1] 99.594 99.990 71.280 89.793 39.303# Función para visualizar la distribution beta
plot_beta <- function(alpha, beta) {
x <- seq(0, 1, length.out = 100)
y <- dbeta(x, alpha, beta)
data <- data.frame(x = x, y = y)
ggplot(data, aes(x, y)) +
geom_line()
}
# Distirbuciones beta
Latentes <- plot_beta(0.396, 99.594) +
xlim(0, .1) +
labs(
title = paste("Distribución de latentes \n (alpha =", 0.396, ", beta =", 99.594, ")"),
x = "Probabilidad", y = "Densidad"
) +
theme(text = element_text(size = 6)) +
geom_vline(xintercept = 0.004, linetype = "dashed", color = "#009392")
Plantulas <- plot_beta(0.000, 99.990) +
xlim(0, .025) + ylim(0, .05) +
labs(
title = paste("Distribución de plántulas \n (alpha =", 0.000, ", beta =", 99.90, ")"),
x = "probabilidad", y = "Densidad"
) +
theme(text = element_text(size = 6)) +
geom_vline(xintercept = 0.000, linetype = "dashed", color = "#009392")
Rosetta <- plot_beta(28.710, 71.280) +
xlim(0, .5) +
labs(
title = paste("Distribución de rosettas \n (alpha =", 28.710, ", beta =", 71.280, ")"),
x = "probabilidad", y = "Densidad"
) +
theme(text = element_text(size = 6)) +
geom_vline(xintercept = 0.287, linetype = "dashed", color = "#009392")
Adultos <- plot_beta(10.197, 89.793) +
xlim(0, .27) +
labs(
title = paste("Distribución de Ind. con flores \n (alpha =", 10.197, ", beta =", 89.793, ")"),
x = "probabilidad", y = "Densidad"
) +
theme(text = element_text(size = 6)) +
geom_vline(xintercept = 0.102, linetype = "dashed", color = "#009392")
Fallecidos <- plot_beta(60.687, 39.303) +
xlim(.40, .8) +
labs(
title = paste("Distribución de fallecidos \n (alpha =", 60.687, ", beta =", 39.303, ")"),
x = "probabilidad", y = "Densidad"
) +
theme(text = element_text(size = 6)) +
geom_vline(xintercept = 0.607, linetype = "dashed", color = "#009392")
ggarrange(Latentes, Plantulas, Rosetta, Adultos, Fallecidos, ncol = 3, nrow = 2)
ggsave("images/distribucion_beta_Serapias_A3.png")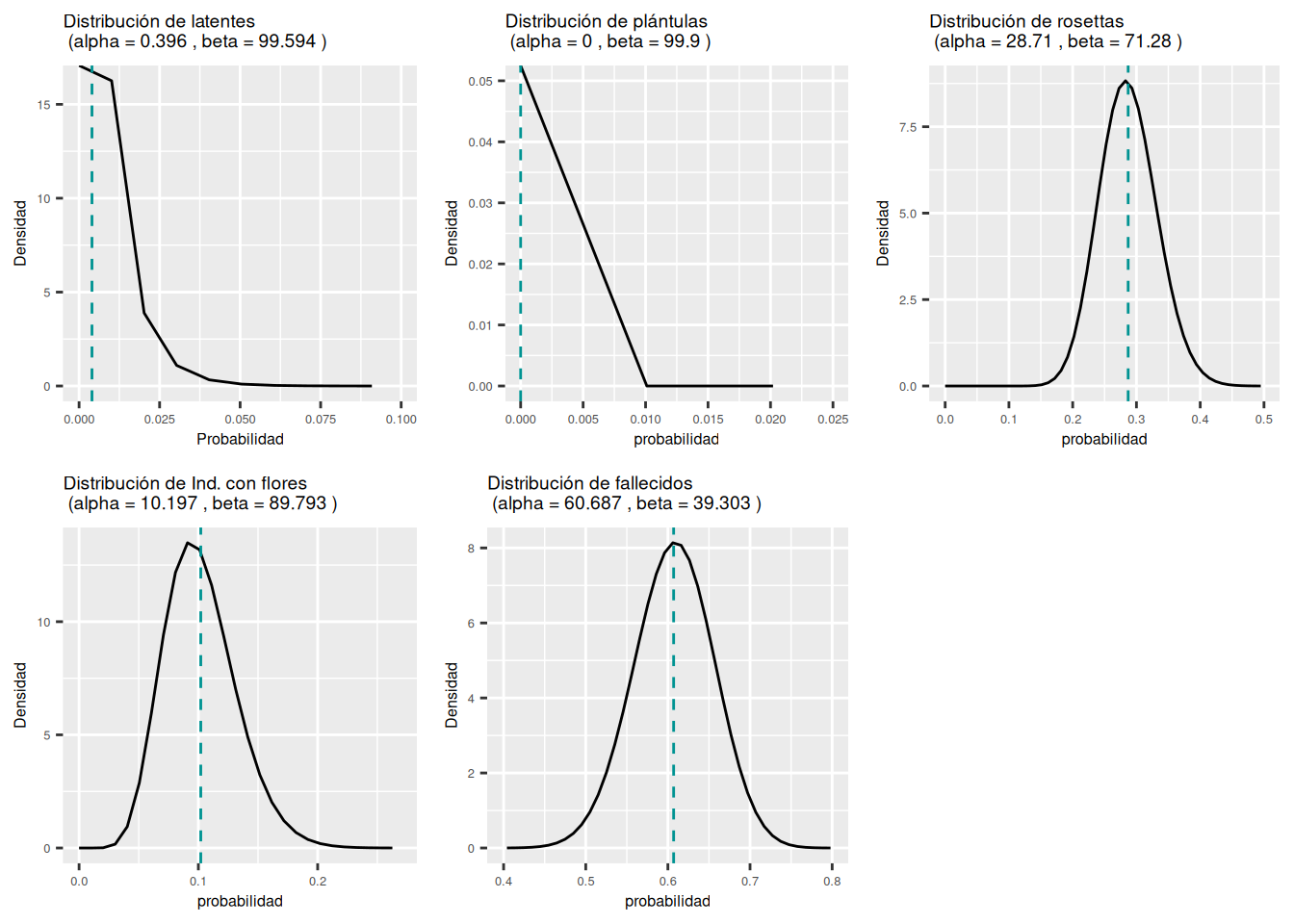
La primera ventaja de usar métodos de análisis por simulaciones es que se pueden calcular intervalos de confianza. Si los intervalos de confianza de lambda solapan el uno (1) (o solapan el 0 para \(\log\lambda\)) entonces no hay evidencia de que el crecimiento poblacional sea diferente de estable.
La segunda ventaja de usar simulaciones es que hay que recordar que los análisis están basado en datos limitados y que si hubiese más o menos datos para estimar los elementos de la matriz, los parámetros de la matriz pudiesen ser diferentes. Entonces hay un componente de incertidumbre en los estimados de los parámetros de la matriz, los valores de promedio no recolectan esa variación entre individuos, tiempo y espacio.
La incertidumbre por causas intrínsecas (los individuos, la genética, el tamaño de muestra) y extrínsecas (hábitat, tiempo, variación en polinizadores, herbívoros, etc.) se evalúa usando análisis de variación espacial, temporal y demográfica. La incertidumbre en los parámetros de la matriz de transición se puede cuantificar con intervalos de confianza o con el intervalo de credibilidad (concepto bayesiano). Es de suma importancia que se evalúe la incertidumbre en los parámetros de la matriz de transición para tener un mejor entendimiento de dónde proviene la variación y la incertidumbre en el crecimiento poblacional de una especie, y saber si hay suficiente evidencia para tener confianza en si la especie está en declive, crece o se mantiene estable.